 Feedback
FeedbackThis section provides the PICO questions posed, the data and studies considered to answer the questions, the methods used for analysis and data synthesis, a summary of evidence on desirable and undesirable effects and certainty of evidence, and a summary of other evidence considered during development of the recommendation. Additional detail on the evidence is available in the web annexes containing the GRADE evidence summary tables (Web Annex 3) and GRADE evidence-to-decision tables (Web Annex 4).
PICO questions
The recommendation in this section is a result of assessments of the PICO questions listed below. Because of the different intervention and comparator groups used, PICOs 3, 5 and 6 have been split into several sub-PICO questions (details are given in the text and in Table 1.3).
PICO question 3–2022 (MDR/RR-TB, 2022): Should BPaL regimens with lower linezolid exposure (dose or duration) be used instead of the original BPaL regimen in patients who are eligible for BPaL regimen?
PICO question 4–2022 (MDR/RR-TB, 2022): Should 6-month regimen using bedaquiline, pretomanid, linezolid be used in patients with pulmonary pre-XDR-TB (MDR/RR-TB with fluoroquinolone resistance)?
PICO question 5–2022 (MDR/RR-TB, 2022): Should 6-month regimen using bedaquiline, pretomanid and linezolid be used in patients with pulmonary MDR/RR-TB and without fluoroquinolone resistance?
PICO question 6–2022 (MDR/RR-TB, 2022): Should 6-month regimen using bedaquiline, pretomanid and linezolid with or without addition of moxifloxacin (BPaLM) or clofazimine be used in patients with pulmonary MDR/RR-TB (with or without fluoroquinolone resistance)?
Data and studies considered
The review of this group of PICO questions during the GDG meeting convened by WHO in February–March 2022 was based on new evidence provided by MSF from the TB-PRACTECAL clinical trial and by the TB Alliance from the ZeNix trial. For several assessments under this PICO question, the data from the 2021 WHO individual patient dataset (IPD) were used. Patient populations included in two trials were recruited following strict inclusion and exclusion criteria; the populations had many similarities and few notable differences. The highlights of the criteria used by these trials are presented in Table 1.1. For a complete list of the exclusion criteria, see Annex 2 and published trial protocols.14
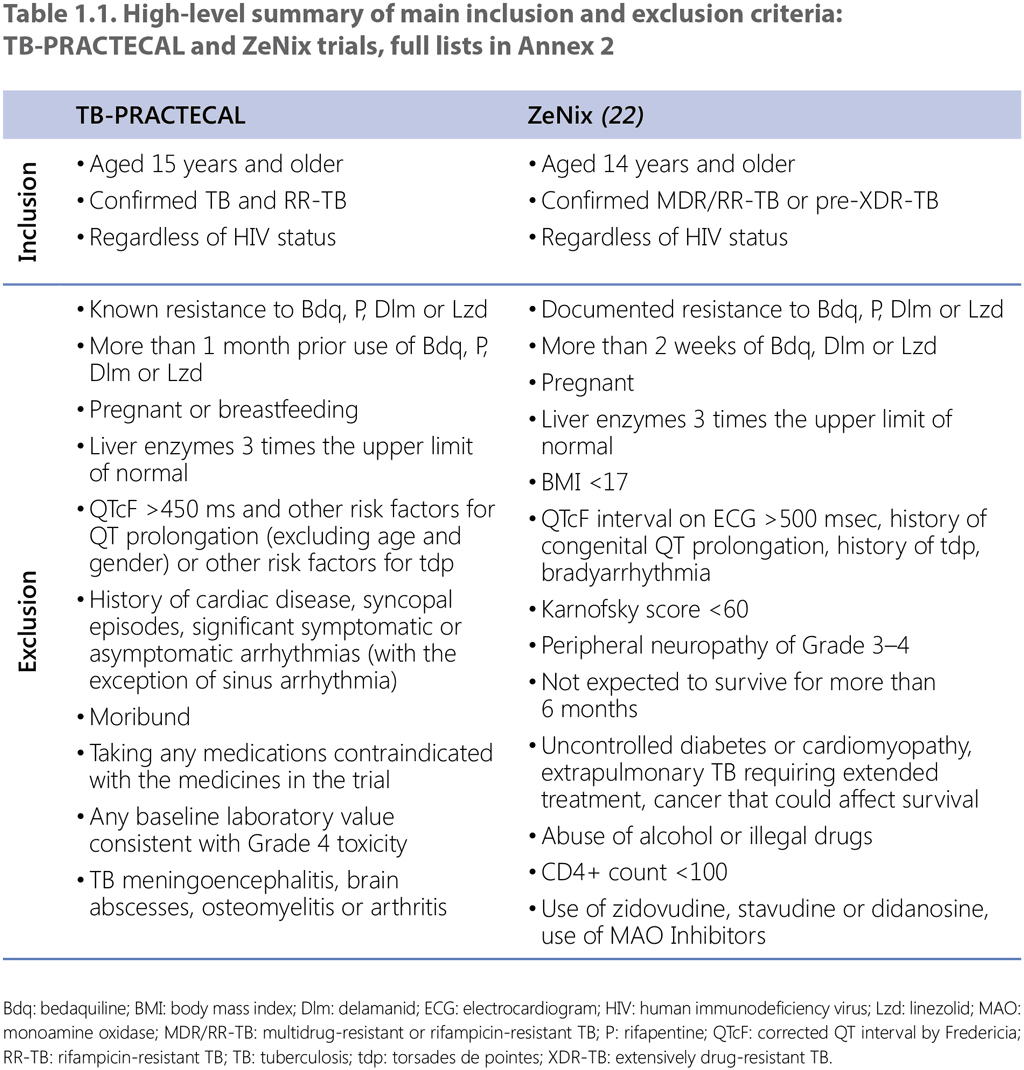
TB-PRACTECAL
TB-PRACTECAL was a multicentre, open-label, multi-arm, randomized, controlled, multistage, Phase 2–3 trial evaluating short treatment regimens containing bedaquiline and pretomanid in combination with existing and repurposed anti-TB drugs (e.g. linezolid and clofazimine) for the treatment of microbiologically confirmed pulmonary MDR/RR-TB.15
The study was divided into two stages, with a seamless transition between the stages, meaning that recruitment into an arm would only stop after a decision had been taken following stage 1 primary endpoint data analysis. In the first stage – equivalent to a Phase 2B trial of safety and preliminary efficacy – patients were randomly assigned one of four regimens, stratified by site. Investigational regimens included oral bedaquiline, pretomanid and linezolid. Two of the regimens also included moxifloxacin (arm 1) and clofazimine (arm 2). The main objective of Stage 1 was to select drug regimens for evaluation in stage 2, based on 8-week safety and efficacy endpoints. Investigational arms that did not meet predefined safety and efficacy criteria were not considered for further evaluation.
The second stage of the study was equivalent to a Phase 3 trial investigating the safety and efficacy of the most promising regimen. As intended in the study protocol, the regimen was evaluated for safety and efficacy in comparison with the SoC arm at 72 weeks after randomization. Stage 2 of the trial included an intervention arm of BPaLM compared with the locally approved SoC, consistent with WHO recommendations for the treatment of MDR/RR-TB or pre-XDR-TB at the time of trial conduct (including a 9–12-month injectable-containing regimen; 18–24-month WHO-recommended regimen [pre-2019]; 9–12-month all-oral regimen; and 18–20-month all-oral regimen). The TB-PRACTECAL trial stopped enrolling patients soon after its independent data safety and monitoring board indicated that the BPaLM regimen is superior to the SoC, because it was considered that more data were extremely unlikely to change the results of the trial. This trial was not designed to compare the investigational regimens against each other.
Eligible patients were aged 15 years and older, and had bacteriologically (molecular or phenotypic) confirmed TB and resistance to at least rifampicin by a molecular or phenotypic drug susceptibility test. The primary efficacy outcome was the composite endpoint of unfavourable outcomes (failure, death, treatment discontinuation, recurrence or loss to follow-up) at 72 weeks after randomization. Relevant secondary efficacy outcomes included culture conversion at 12 and 24 weeks, unfavourable outcomes at 24 weeks after randomization, unfavourable outcomes at 108 weeks after randomization, median time to culture conversion and recurrence by week 48 in the investigational arms. Participants were randomized in a 1:1:1:1 ratio into either the SoC or one of the following three intervention arms:
- Arm 1: 24 weeks of B-Pa-Lzd-Mfx (BPaLM);
- Arm 2: 24 weeks of B-Pa-Lzd-Cfz (BPaLC); and
- Arm 3: 24 weeks of B-Pa-Lzd (BPaL).
In all intervention arms, linezolid was given at 600 mg daily for 16 weeks then 300 mg daily for the remaining 8 weeks (or earlier when moderately tolerated). Bedaquiline was given at 400 mg once daily for 2 weeks followed by 200 mg three times per week for 22 weeks. Safety monitoring for most participants included multiple electrocardiograms (ECGs) at baseline, then weekly until week 8, every 4 weeks up to week 24 and then every 8 weeks thereafter. Microbiological monitoring included smear microscopy and culture at baseline and day 7, then every 4 weeks up until week 24 and every 8 weeks thereafter.
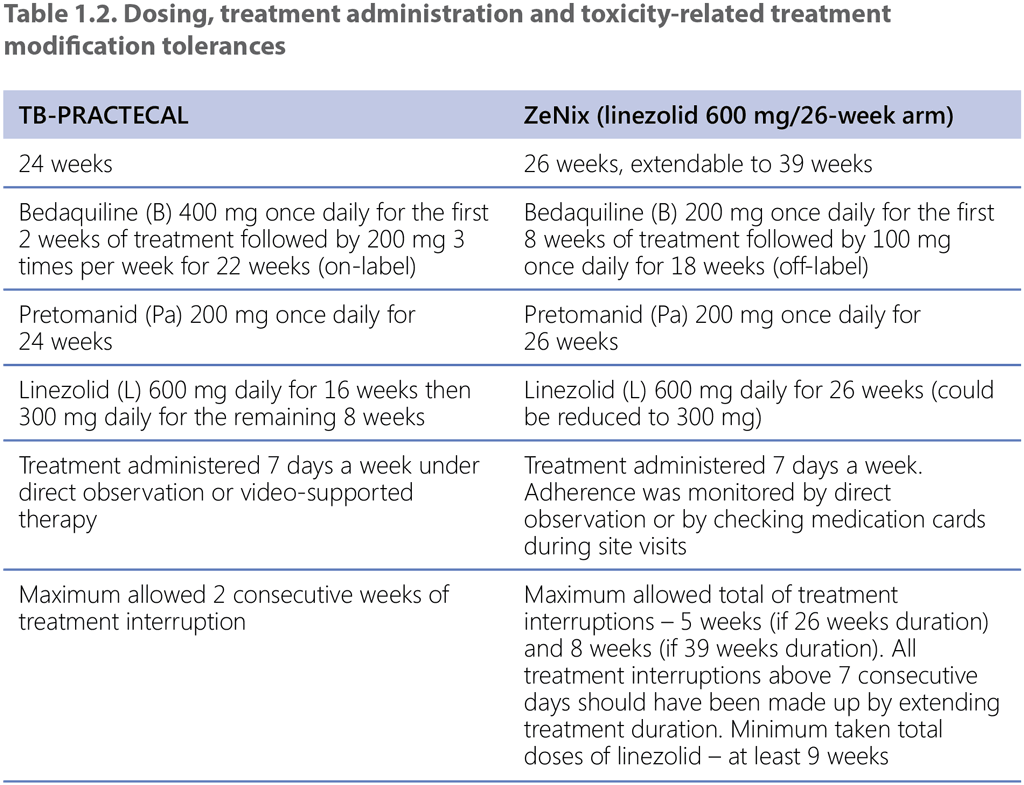
ZeNix
ZeNix was a Phase 3 partially blinded, randomized trial assessing the safety and efficacy of various doses and treatment durations of linezolid plus bedaquiline and pretomanid in individuals with pulmonary MDR/RR-TB and additional resistance to fluoroquinolones (with or without resistance to injectable agents) or those with treatment intolerant or nonresponsive MDR/RR-TB. Eligible patients were aged 14 years and older, weighed at least 35 kg, had a documented HIV result and had bacteriologically confirmed sputum culture positive XDR-TB (pre-2021 definition) or bacteriologically confirmed MDR/RR-TB, but were treatment intolerant or nonresponsive to previous MDR/RR-TB treatment. The primary study outcome was the incidence of bacteriological failure or relapse or clinical failure through follow-up until 26 weeks after the end of treatment. The secondary outcomes included incidence of bacteriological failure or relapse or clinical failure through follow-up until 78 weeks after the end of treatment. Participants received 26 weeks of treatment with BPaL. Each of the four arms varied the dose and duration of linezolid: 1 200 mg 26 weeks, 1 200 mg 9 weeks, 600 mg 26 weeks or 600 mg 9 weeks. Bedaquiline was given at 200 mg once daily for 8 weeks then 100 mg once daily for 18 weeks. This off-label dosing schedule is supported by pharmacokinetic simulations for an alternative bedaquiline dosing schedule that provides comparable exposures and was developed to support adherence and facilitate treatment administration (all medicines daily throughout the regimen) (23).
Safety monitoring included scheduled testing and assessments of laboratory parameters, ECG, vital signs and other physical examinations (24). Microbiological monitoring included smear microscopy, molecular testing and liquid culture from sputum at baseline and liquid culture at all patient visits thereafter (24).

2021 WHO IPD
In 2021, WHO issued a public call for data to serve as a comparator group (SoC) against which 6–9-month regimens could be compared. These cohorts received treatment conforming to the WHO DR-TB guidelines of 2020 with bedaquiline and linezolid for a duration ranging from 6 to 24 months. Patients receiving injectable antibiotics were excluded.
Included datasets comprised individuals using one of the following regimens:
- 6–12-month all-oral regimens using at least bedaquiline and linezolid; or
- 9–12-month WHO (2019) all-oral bedaquiline-containing regimen in the combination, such as 4–6 Bdq(6m)-Lfx/Mfx-Cfz-Z-E-Hh-Eto / 5m Lfx/Mfx-Cfz-Z-E; or
- ≥18-month WHO (2018) all-oral treatment regimen containing at least bedaquiline and linezolid.
The individual datasets that are included in this cohort are described in detail in the statistical analysis plan (Web Annex 6). To be eligible for inclusion in a short comparator regimen (target 9–12 months at treatment commencement), patients must have fulfilled each of the following:
- had a treatment duration not exceeding 12 months;
- received six or more drugs during treatment, including bedaquiline; and
- if given an outcome of cure or completed, had a treatment duration of 8.5 months or more.
To be eligible for inclusion in a longer comparator regimen (target 18–24 months), patients must have fulfilled each of the following:
- be classified in the dataset as having received a longer regimen (if stated);
- had a treatment duration not longer than 24 months;
- received four or more drugs (regardless of drug susceptibility; i.e. regardless of whether they were likely to be effective), including bedaquiline; and
- if given an outcome of cure or complete, had a treatment duration of 17.5 months or more.
Methods used for analysis and data synthesis
Descriptive analyses of the baseline characteristics of participants in all included studies were performed; characteristics included demographics, diagnostic test results, treatment regimens and treatment outcomes.
Comparative analyses were performed within individual studies and between multiple studies:
- Within study comparisons – for studies in which both a short-course (6 months in duration) regimen and a relevant comparator are used, pairwise comparisons were conducted between each of the short-course regimens and the comparator. For included RCTs (e.g. the TB-PRACTECAL trial and NExT trial), the primary outcome of the prespecified analysis was also calculated and reported.
- Pairwise comparisons between studies – comparisons addressing each PICO question were conducted by comparing outcomes among cohorts in which participants received either the intervention or the control regimen relevant to that question.
Statistical models
For comparisons between dataset or cohorts, outcomes were presented as unadjusted and adjusted risk ratios (RR). Adjusted risk ratios (aRR) were calculated using a log-binomial generalized linear regression (binomial error distribution with log link function). Pre-specified potential confounders were adjusted for using inverse probability propensity score weighting. No convergence issues arose with the log-binomial model. When outcome rates were close to the boundary, aRR were not calculated, and unadjusted RR were presented. For outcomes where the number of outcome events was zero, an unadjusted risk difference (RD) was calculated. For unadjusted RDs or RRs, 95% confidence intervals (CIs) were calculated using the score method. Covariate selection for calculation of propensity scores was based on data availability and clinical knowledge. The covariates considered for inclusion in the propensity scores analysis included age, gender, baseline smear result, HIV status (including ART status), prior treatment history (including whether previous infection was drug resistant), body mass index (BMI), smoking status, diabetes diagnosis, cavitation at baseline, presence of bilateral disease and fluroquinolone resistance. For the calculation of aRRs, multiple imputation by chain equations using the “within” propensity score approach was used to account for missing data in potential confounders when the proportion of missing values for a confounder was less than 45%.
Timing of follow-up for comparisons between regimens
The analyses undertaken for this evidence review combined results from cohorts with differing follow-up times after initiation of treatment. There were differences in the follow-up time between cohorts (from 5.5 months to 24 months) and within single cohorts (e.g. the WHO IPD 2021 dataset combined multiple cohorts with variable follow-up times). Follow-up time was separated into the time between commencement of treatment and treatment completion, and the period from treatment completion until the end of follow-up. For shorter regimens, post-treatment follow-up was particularly important because higher relapse rates may be a consequence of shorter treatments that do not completely remove M. tuberculosis. Where possible, it was important for follow-up time between two groups in a comparison to be equivalent, so that participants had an equivalent likelihood of death or relapse. In these analyses, the follow-up time was measured from the start date of treatment rather than after the date of treatment completion, to minimize the effect of differences in total follow-up time.
The principles for accounting for time periods of follow-up were as follows:
- Where possible, follow up participants in the intervention and control groups for the same total time, so that the likelihood of unsuccessful outcomes (e.g. death) is the same in both groups.
- Limit follow-up to 24 months after treatment initiation for all cohorts. There were no analyses in which both intervention and comparator cohorts had more than 24 months of follow-up available. The evidence accumulated from TB treatment trials demonstrates that a high proportion of recurrences are likely to occur within 12 or even 6 months of stopping treatment (25).
- ≥Select a primary analysis that optimizes the number of participants included in both groups. For shorter (6–9-month regimens), follow-up time in the comparison was included to allow for relapse to be captured.
Additional sensitivity analyses were performed, where possible, evaluating the effect of follow-up time upon treatment outcomes.
Summary of evidence on desirable and undesirable effects and certainty of evidence
The evidence on the novel regimens to inform PICO questions was derived from two trials. It included information on a total of 419 of 423 participants who were enrolled in four arms of the TB-PRACTECAL and on 172 of 181 participants who were enrolled in four arms of the ZeNix trial16.
Data from patients in relevant arms of these trials were used in each of the comparisons that led to the conclusions and final recommendation on the use of the BPaLM/BPaL regimen. Even though the TB-PRACTECAL trial was not designed to compare the investigational regimens against each other and with the SoC, the comparisons of the different arms of the trial to the BPaLM arm (sub-PICOs 6.2 to 6.6) were performed to aid the panel in making final decisions.
Sub-PICO 3.2
The BPaL 1200–9 arm of the ZeNix trial (where linezolid 1 200 mg daily was used for 9 weeks) was compared with the BPaL 1200–26 arm (where linezolid 1 200 mg daily was used for 26 weeks) in the same population of patients with MDR/RR-TB with or without fluoroquinolone resistance. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 1200–9 (n=43) compared with participants with the same resistance patterns receiving BPaL with linezolid 1200–26 (n=44) experienced:
- lower levels of treatment success (93% vs 98%); that is, a 5% relative reduction (RR=0.95, 95% CI: 0.87 to 1.05);
- higher levels of failure and recurrence (4.7% vs 2.3%); that is, a twofold relative increase (RR=2.1, 95% CI: 0.19 to 22);
- higher levels of deaths (2.3% vs 0%); that is, a 2% absolute increase (RD=0.02, 95% CI: –0.06 to 0.12);
- the same levels of loss to follow-up (0% vs 0%); that is, a 0% absolute difference (RD=0.00, 95% CI: –0.08 to 0.08);
- lower levels of adverse events (16% vs 18%); that is, a 10% relative reduction (RR=0.90, 95% CI: 0.36 to 2.3); and
- the same levels of amplification of drug resistance (0% vs 0%); that is, a 0% absolute difference (RD=0.00, 95% CI: –0.08 to 0.08).
The GDG judged the benefits of BPaL with linezolid 1200–9 to be small and the undesirable effects to be moderate compared with BPaL with linezolid 1200–26. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 1200–26.
Conclusion
The use of the 26 weeks of 1 200 mg linezolid is suggested over 9 weeks of 1 200 mg linezolid as part of the BPaL regimen in adults with MDR/RR-TB or pre-XDR-TB.
Sub-PICO 3.3
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks) was compared with the BPaL 1200–26 arm (where linezolid 1 200 mg daily was used for 26 weeks) in the same population of patients with MDR/RR-TB with or without fluoroquinolone resistance. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 600–26 (n=43) compared with participants with the same resistance patterns receiving BPaL with linezolid 1200–26 (n=44) experienced:
- higher levels of treatment success (100% vs 98%); that is, a 2% relative increase (RR=1.02, 95% CI: 0.98 to 1.07);
- lower levels of failure and recurrence (0% vs 2.3%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.12 to 0.06);
- lower levels of Grade 3–5 adverse events (14% vs 18.6%); that is, a 23% relative reduction (RR=0.77, 95% CI: 0.29 to 2.03); and
- the same levels of deaths (0% vs 0%), loss to follow-up (0% vs 0%) or amplified resistance (0% vs 0%).
The GDG judged the benefits of BPaL with linezolid 600–26 to be moderate and the undesirable effects to be trivial compared with BPaL with linezolid 1200–26. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 26 weeks of 600 mg linezolid over 26 weeks of 1 200 mg linezolid is suggested as part of the BPaL regimen in adults with MDR/RR-TB or pre-XDR-TB.
Sub-PICO 3.4
The BPaL 600–9 arm of the ZeNix trial (where linezolid 600 mg daily was used for 9 weeks) was compared with the BPaL 1200–26 arm (where linezolid 1 200 mg daily was used for 26 weeks) in the same population of patients with MDR/RR-TB with or without fluoroquinolone resistance. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 600–9 (n=42) compared with participants with the same resistance patterns receiving BPaL with linezolid 1200–26 (n=44) experienced:
- lower levels of treatment success (93% vs 98%); that is, a 5% relative reduction (RR=0.95, 95% CI: 0.86 to 1.05);
- higher levels of failure and recurrence (4.8% vs 2.3%); that is, a twofold increase (RR=2.10, 95% CI: 0.20 to 22.26);
- higher levels of loss to follow-up (2.4% vs 0%); that is, a 2% absolute increase (RD=0.02, 95% CI: –0.06 to 0.12);
- lower levels of Grade 3–5 adverse events (14.3% vs 18.2%); that is, a 21% relative reduction (RR=0.79, 95% CI: 0.30 to 2.07); and
- the same levels of deaths (0% vs 0%) or amplified resistance (0% vs 0%).
The GDG judged the benefits of BPaL with linezolid 600–9 to be small and the undesirable effects to be moderate compared with the BPaL with linezolid 1200–26. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 1200–26.
Conclusion
The use of the 26 weeks of 1 200 mg over 9 weeks of 600 mg linezolid is suggested as part of the BPaL regimen in adults with MDR/RR-TB or pre-XDR-TB.
PICO 3 – Intermediate summary conclusion
The assessment of PICO 3 allowed for the decision on the optimal dosing and duration of linezolid within the BPaLM/BPaL regimen, and narrowed down the subsequent comparisons to the intervention regimen with this particular dose and duration of linezolid – BPaL (600 mg – 26 weeks).
Sub-PICO 4.1
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients with fluoroquinolone resistance from the 2021 IPD who were receiving longer regimens for treatment of MDR/RR-TB, designed in line with 2020 WHO guidelines. Primary analysis was undertaken at 18 months post treatment initiation.
Participants with pulmonary pre-XDR-TB (MDR/RR-TB with fluoroquinolone resistance) receiving BPaL 600–26 (n=33) compared with participants receiving longer regimens for MDR/RR-TB (n=839) experienced:
- higher levels of treatment success (100% vs 75%); that is, a 34% relative increase (RR=1.34, 95% CI: 1.20 to 1.40);
- lower levels of failure and recurrence (0% vs 6.6%); that is, a 7% absolute reduction (RD= –0.07, 95% CI: –0.08 to –0.04);
- lower levels of deaths (0% vs 9.9%); that is, a 10% absolute reduction (RD= –0.10, 95% CI: –0.12 to –0.01);
- lower levels of loss to follow-up (0% vs 9.1%); that is, a 9% absolute reduction (RD= –0.09, 95% CI: –0.11 to –0.01); higher levels of adverse events (15% vs 4.4%); that is, a 3.4-fold increase (RR=3.44, 95% CI: 1.44 to 8.17); and
- lower levels of amplification of drug resistance (0% vs 7.4%); that is, a 7% absolute reduction (RD= –0.07, 95% CI: –0.09 to –0.03).
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with longer regimens recommended by WHO. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than a longer (18-month) regimen is suggested in patients with MDR/RR-TB and resistance to fluoroquinolones (pre-XDR-TB), who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
PICO 4 – Intermediate conclusion
The assessment of PICO 4 resulted in the conditional recommendation for use of the BPaL (600 mg – 26 weeks) regimen over the currently recommended longer regimens in patients with MDR/RR-TB and additional fluoroquinolone resistance (pre-XDR-TB).
Sub-PICO 5.1
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with or without fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients without fluoroquinolone resistance treated in South Africa with the WHO-recommended 9-month regimen with ethionamide for 4 months. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaL 600–26 regimen (n=43) compared with participants with MDR/RR-TB (without fluoroquinolone resistance) receiving the 9-month regimen with ethionamide (n=785) experienced:
- higher levels of treatment success (100% vs 69%); that is, a 45% relative increase (RR=1.45, 95% CI: 1.32 to 1.53);
- lower levels of failure and recurrence (0% vs 1.3%); that is, a 1% absolute reduction (RD=–0.01, 95% CI: –0.02 to 0.07);
- lower levels of deaths (0% vs 19%); that is, a 19% absolute reduction (RD=–0.19, 95% CI: –0.22 to –0.1);
- lower levels of loss to follow-up (0% vs 11%); that is, an 11% absolute reduction (RD= –0.11, 95% CI: –0.14 to –0.03); and
- the same levels of amplified resistance (0% vs 0%); that is, a 0% absolute difference (RD= 0.00, 95% CI: –0.01 to 0.08).
Grade 3–5 adverse events were noted in 14% of participants receiving the BPaL 600–26 but no comparison could be done because no data were available for participants receiving the 9-month regimen with ethionamide.
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with the WHO-recommended 9-month regimen with ethionamide. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than the 9-month regimen (with ethionamide) is suggested in patients with MDR/RR-TB without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 5.2
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with or without fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients without fluoroquinolone resistance from the 2021 IPD, treated with longer regimens for MDR/RR-TB, designed in line with the 2020 WHO guidelines. Primary analysis was undertaken at 18 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL 600–26 regimen (n=43) compared with participants with MDR/RR-TB (without fluoroquinolone resistance) receiving longer regimens recommended by WHO (n=850) experienced:
- higher levels of treatment success (98% vs 74%); that is, a 32% relative increase (RR=1.32, 95% CI: 1.19 to 1.39);
- lower levels of failure and recurrence (2.3% vs 3.3%); that is, a 29% relative reduction (RR=0.71, 95% CI: 0.12 to 3.8);
- lower levels of deaths (0% vs 11%); that is, an 11% absolute reduction (RD= –0.11, 95% CI: –0.13 to –0.03);
- lower levels of loss to follow-up (0% vs 12%); that is, a 12% absolute reduction (RD= –0.12, 95% CI: –0.14 to –0.04);
- higher levels of Grade 3–5 adverse events (14% vs 5%); that is, a fourfold relative increase (aRR=3.99, 95% CI: 1.67 to 9.57); and
- lower levels of amplified resistance (0% vs 2.4%); that is, a 2% absolute decrease (RD= –0.02, 95% CI: –0.04 to 0.06).
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with longer regimens recommended by WHO. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than longer (18-month) regimens is suggested in patients with MDR/RR-TB and without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 5.3
The BPaL 600–26 arm of the ZeNix trial (where linezolid 600 mg daily was used for 26 weeks and the population included patients with MDR/RR-TB with or without fluoroquinolone resistance) was compared with a cohort of MDR/RR-TB patients without fluoroquinolone resistance treated in South Africa with a 9-month regimen with linezolid for 2 months. Primary analysis was undertaken at 12 months post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL with linezolid 600–26 (n=43) compared with participants with MDR/RR-TB (without fluoroquinolone resistance) receiving a 9-month regimen with linezolid (n=4 216) experienced:
- higher levels of treatment success (100% vs 66%); that is, a 52% relative increase (RR=1.52, 95% CI: 1.38 to 1.55);
- lower levels of failure and recurrence (0% vs 1.2%); that is, a 1% absolute reduction (RD= –0.01, 95% CI: –0.02 to 0.07);
- lower levels of deaths (0% vs 18%); that is, an 18% absolute reduction (RD= –0.18, 95% CI: –0.19 to –0.1);
- lower levels of loss to follow-up (0% vs 15%); that is, a 15% absolute reduction (RD= –0.15, 95% CI: –0.16 to –0.07);
- higher levels of Grade 3–5 adverse events (14% vs 4.9%); that is, a threefold increase (aRR=2.92, 95% CI: 1.38 to 6.18); and
- lower levels of amplified resistance (0% vs 0.6%); that is, a 1% absolute reduction (RD= –0.01, 95% CI: –0.01 to 0.08).
The GDG judged the benefits of BPaL with linezolid 600–26 to be large and the undesirable effects to be moderate compared with receiving a 9-month regimen with linezolid. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaL with linezolid 600–26.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than the 9-month regimen (with linezolid) is suggested in patients with MDR/RR-TB without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
PICO 5 – Intermediate summary conclusion
The three assessments performed under PICO 5 resulted in the conditional recommendations for the BPaL (600 mg – 26 weeks) regimen over the currently recommended 9-month regimen with ethionamide (sub-PICO 5.1), over longer (18-month) regimens (sub-PICO 5.2) and over the new 9-month regimen where ethionamide is replaced with 2 months of linezolid (sub-PICO 5.3) in patients with pulmonary MDR/RR-TB without fluoroquinolone resistance.
Sub-PICO 6.1
The BPaLM regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the comparator arm of the TB-PRACTECAL trial, which comprised MDR/RR-TB or pre-XDR-TB patients treated with multiple local SoC regimens recommended by WHO at the time the trial was conducted (including a 9–12-month injectable-containing regimen, an 18–24-month WHO-recommended regimen [pre-2019], a 9–12-month all-oral regimen and an 18–20-month all-oral regimen). Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaLM regimen (n=62) compared with participants receiving WHO-recommended SoC regimens used in the TB-PRACTECAL trial (n=66) experienced:
- higher levels of treatment success (89% vs 52%); that is, a 73% relative increase (aRR=1.73, 95% CI: 1.31 to 2.27);
- lower levels of failure and recurrence (8% vs 26%) that is 74% relative reduction (aRR=0.26, 95%CI 0.10 to 0.71);
- lower levels of deaths (0% vs 3.0%); that is, a 3% absolute reduction (RD= –0.03, 95% CI: –0.10 to 0.03);
- lower levels of loss to follow-up (3.2% vs 20%); that is, a 84% relative reduction (RR=0.16, 95% CI: 0.04 to 0.61);
- lower levels of Grade 3–5 adverse events (21% vs 51%); that is, a 59% relative reduction (aRR=0.41, 95% CI: 0.26 to 0.63); and
- lower levels of amplified resistance (0% vs 1.9%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.07 to 0.02).
The GDG judged the benefits of BPaLM to be large and the undesirable effects to be trivial compared with WHO-recommended SoC regimens. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours the BPaLM regimen.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) rather than a 9-month or longer (18-month) regimen is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.2
The BPaLM regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the BPaL arm of the TB-PRACTECAL trial, which comprised MDR/RR-TB or pre-XDR-TB patients. Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaLM regimen (n=62) compared with participants receiving BPaL in the TB-PRACTECAL trial (n=60) experienced:
- higher levels of treatment success (89% vs 77%); that is, a 15% relative increase (aRR=1.15, 95% CI: 0.95 to 1.38);
- lower levels of failure and recurrence (8.1% vs 13%); that is, a 47% relative reduction (aRR= 0.53, 95% CI: 0.17 to 1.63);
- lower levels of loss to follow-up (3.2% vs 10%); that is, a 68% relative reduction (aRR=0.32, 95% CI: 0.08 to 1.34);
- no difference in deaths (0% vs 0%); that is, a 0% absolute difference (RD= 0, 95% CI: –0.06 to 0.06);
- higher levels of Grade 3–5 adverse events (21% vs 20%); that is, a 7% relative increase (aRR=1.07, 95% CI: 0.62 to 1.88); and
- lower levels of amplified resistance (0% vs 2.9%); that is, a 3% absolute reduction (RD= –0.03, 95% CI: –0.08 to 0.01).
The GDG judged the benefits of BPaLM to be moderate and the undesirable effects to be small compared with BPaL. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaLM.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) rather than BPaL is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.3
The BPaLM regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the BPaLC arm of the TB-PRACTECAL trial that comprised MDR/RR-TB or pre-XDR-TB patients. Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving the BPaLM regimen (n=62) compared with participants receiving the BPaLC regimen (n=64) in the TB-PRACTECAL trial experienced:
- higher levels of treatment success (89% vs 81%); that is, an 11% relative increase (aRR 1.11, 95% CI: 0.94 to 1.31);
- lower levels of failure and recurrence (8.1% vs 9.4%); that is, a 30% relative reduction (aRR= 0.70, 95% CI: 0.2 to 2.29);
- lower levels of deaths (0% vs 1.6%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.08 to 0.04);
- lower levels of loss to follow-up (3.2% vs 7.8%); that is, a 59% relative reduction (RR=0.41, 95% CI: 0.09 to 1.77);
- lower levels of Grade 3–5 adverse events (21% vs 34%); that is, a 39% relative reduction (aRR=0.61, 95% CI: 0.37 to 1.00); and
- lower levels of amplified resistance (0% vs 1.9%); that is, a 2% absolute reduction (RD= –0.02, 95% CI: –0.07 to 0.02).
The GDG judged the benefits of BPaLM to be moderate and the undesirable effects to be trivial compared with BPaLC. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaLM.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and moxifloxacin (BPaLM) rather than BPaLC is suggested in patients with MDR/RR-TB with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.4
The BPaLC regimen arm of the TB-PRACTECAL trial with population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared to the comparator arm of the TB-PRACTECAL trial comprised of MDR/RR-TB or pre-XDR-TB patients treated with multiple local SoC regimens recommended by WHO at the time of trial conduct (including a 9–12-month injectable-containing regimen; 18–24-month WHO-recommended regimen [pre-2019]; 9–12-month all-oral regimen; and 18–20-month all-oral regimen). Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaLC (n=64) compared to participants receiving WHO-recommended SoC regimens used in the TB-PRACTECAL trial (n=66) experienced:
- higher treatment success (81% vs 52%); that is, a 55% relative increase (aRR=1.55, 95% CI: 1.15 to 2.11);
- lower levels of failure and recurrence (9.4% vs 26%); that is, a 66% relative reduction (aRR=0.34, 95% CI: 0.14 to 0.87);
- lower levels of deaths (1.6% vs 3.0%); that is, a 48% relative reduction (RR=0.52, 95% CI: 0.07 to 3.85);
- lower levels of loss to follow-up (7.8% vs 20%); that is, a 57% relative reduction (aRR=0.43, 95% CI: 0.15 to 1.23);
- lower levels of grade 3 to 5 adverse events (34% vs 51%); that is, a 33% relative reduction (aRR=0.67, 95% CI: 0.46 to 0.97); and
- and higher levels of amplified resistance (1.9% vs 1.9%); that is, a 4% relative increase (RR=1.04, 95% CI: 0.19 to 5.80).
The GDG judged the benefits of BPaLC to be large and the undesirable effects to be trivial compared to WHO-recommended SoC regimens. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours BPaLC.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid, linezolid and clofazimine (BPaLC) rather than a 9-month or longer (18-month) regimen is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month (overruled by conclusions of sub-PICO 6.5 and sub-PICO 6.6).
Sub-PICO 6.5
The BPaLC regimen arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the BPaL arm of the TB-PRACTECAL trial that comprised MDR/RR-TB or pre-XDR-TB patients. Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with pulmonary MDR/RR-TB or pre-XDR-TB receiving BPaLC (n=64) compared with participants receiving BPaL 600–300 (n=60) experienced:
- higher levels of treatment success (81% vs 77%); that is, a 4% relative increase (aRR=1.04, 95% CI: 0.84 to 1.30);
- lower levels of failure and recurrence (9.4% vs 13%); that is, a 14% relative reduction (aRR=0.86, 95% CI: 0.28 to 2.69);
- higher levels of deaths (1.6% vs 0%); that is, a 2% absolute increase (RD=0.02, 95% CI: –0.05 to 0.08);
- lower levels of loss to follow-up (7.8% vs 10%); that is, a 28% relative reduction (aRR=0.72, 95% CI: 0.21 to 2.47);
- higher levels of adverse events (34% vs 20%); that is, a 64% relative increase (aRR=1.64, 95% CI: 0.97 to 2.79); and
- lower levels of amplification of drug resistance (1.9% vs 2.9%); that is, a 35% relative reduction (RR=0.65, 95% CI: 0.13 to 3.21).
The GDG judged both the desirable and the undesirable effects of BPaLC to be small compared with BPaL. The certainty of evidence was judged to be very low. The balance of health effects did not favour either the intervention or the comparator; however, taking into consideration the higher cost of the regimen, increased pill burden, reduced acceptability due to skin discolouration and other potential adverse effects related to clofazimine without noticeable net benefit in terms of health effects, the panel judged against the intervention.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than BPaLC is suggested in MDR/RR-TB patients with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
Sub-PICO 6.6
The BPaL arm of the TB-PRACTECAL trial with a population including patients with MDR/RR-TB with or without fluoroquinolone resistance (MDR/RR-TB or pre-XDR-TB) was compared with the comparator arm of the TB-PRACTECAL trial that comprised MDR/RR-TB or pre-XDR-TB patients treated with multiple local SoC regimens (including a 9–12-month injectable-containing regimen, an 18–24-month WHO regimen [pre-2019], a 9–12-month all-oral regimen and an 18–20-month all-oral regimen). Primary analysis was undertaken at 72 weeks post treatment initiation.
Participants with MDR/RR-TB (with or without fluoroquinolone resistance) receiving BPaL (n=60) compared with participants receiving WHO-recommended SoC regimens used in the TB-PRACTECAL trial (n=66) experienced:
- higher levels of treatment success (77% vs 52%); that is, a 47% relative increase (aRR=1.47, 95% CI: 1.09 to 1.99);
- lower levels of failure and recurrence (13% vs 26%); that is, a 48% relative reduction (aRR=0.52, 95% CI: 0.22 to 1.18);
- lower levels of deaths (0% vs 3.0%); that is, a 3% absolute reduction (RD= –0.03, 95% CI: –0.10 to 0.03);
- lower levels of loss to follow-up (10% vs 20%); that is, a 40% relative reduction (aRR=0.60, 95% CI: 0.24 to 1.56);
- lower levels of adverse events (20% vs 51%); that is, a 62% relative reduction (RR=0.38, 95% CI: 0.24 to 0.60); and
- higher levels of amplification of drug resistance (2.9% vs 1.9%); that is, a 59% relative increase (RR=1.59, 95% CI: 0.32 to 7.84).
The GDG judged the benefits of BPaL to be large and the undesirable effects to be trivial compared with WHO-recommended SoC regimens. The certainty of evidence was judged to be very low. Based on this, the panel determined that the balance of health effects probably favours the BPaL regimen.
Conclusion
The use of the 6-month treatment regimen composed of bedaquiline, pretomanid and linezolid (BPaL) rather than a 9-month or longer (18-month) regimen is suggested in patients with MDR/RR-TB with or without resistance to fluoroquinolones, who have either had no previous exposure to bedaquiline and linezolid or have been exposed for less than 1 month.
PICO 6 – Intermediate summary conclusion
The main assessment that defined the overall decision was that of sub-PICO 6.1, which resulted in the conditional recommendation for use of the BPaLM regimen over the internal mix of SoC regimens conforming to the WHO recommendations on 9-month or longer regimens. The assessments of the investigational regimens against each other and with the SoC in sub-PICOs 6.2–6.6 helped the panel in making final decisions.
Summary of other evidence
Additional data reviewed by the GDG relevant to these PICO questions were a cost–effectiveness analysis, a study on the acceptability and likelihood of implementation of the BPaL regimen, modelled pharmacokinetic data based on the development of a pharmacokinetic toxicodynamic model, and a summary of data on potential reproductive toxicity of pretomanid. No additional research data were available during review of sub-PICO questions 3.2–3.5.
Pharmacokinetic data
Early data from the pharmacokinetics study embedded in the TB-PRACTECAL were presented to the GDG panel in one of the preparatory webinars. The final results of this sub-study were not available at the time of the assessment and could not be fully considered.
The pharmacokinetics of linezolid are highly variable, with efficacy and toxicity dependent on factors such as pathogen susceptibility, drug exposure and the combination of companion drugs. The toxicity of linezolid, especially when used at higher doses and longer durations, is a known phenomenon and various strategies have been suggested to reduce it. However, except for the data available from the ZeNix and TB-PRACTECAL trials, no other strategies have been tested in a trial environment.17
Data on reproductive toxicity of pretomanid
New data on the safety of pretomanid based on hormone evaluations in four clinical trials and a paternity survey were assessed; these data have largely alleviated previous concerns about reproductive toxicities observed in animal studies,18 suggesting that adverse effects on human male fertility are unlikely. A study assessing semen in men undergoing treatment that includes pretomanid is in progress and will address any remaining concerns. Below is a summary of preclinical and clinical data relevant to testicular toxicity of pretomanid:
- rodent toxicology studies – evidence of direct testicular toxicity;
- monkey toxicology studies – no evidence for direct testicular toxicity; abnormal sperm findings considered to be secondary to declining physical condition;
- hormone data from clinical studies – no changes in follicle stimulating hormone (FSH), luteinizing hormone (LH) and inhibin B, consistent with testicular toxicity;
- paternity survey – 44 children fathered by 38 men (12%) who participated in pretomanid studies of 4–6 months treatment duration; and
- semen study – ongoing study evaluating semen in men undergoing pretomanid treatment.
Resources required and cost–effectiveness
Estimated regimen costs (in adults) at Global Drug Facility (GDF) prices19 are about US$ 688 for BPaL (600–26), US$ 716 for BPaLM (600–26), an average of US$ 771 for longer regimens (depends on length and composition) and US$ 535–557 for 9-month regimens. Data from three studies were available on more detailed analyses of resources required and cost–effectiveness; two of these studies compared the BPaL regimen with longer (18-month) regimens (26, 27) and one compared the BPaL, BPaLM and BPaLC regimens with longer (18-month) regimens and with the 9-month regimen with ethionamide (28). The applicability of the results from these studies varied by PICO and sub-PICO question, and the panel noted associated caveats when discussing these results (details available in the GRADE evidence-to-decision tables in Web Annex 4). Overall, based on these three publications, estimates for comparative total cost (drugs and delivery) within country appear to be between 1.4-fold and 6-fold higher (longer regimens) or 1–18% higher (9-month regimens) than for BPaLM/BPaL. Thus, the panel judged that implementation of BPaLM/BPaL would probably to lead to large savings when replacing the longer (18-month) regimens and moderate savings when replacing the 9-month regimens.
The cost–effectiveness study (28) found that, in most settings, BPaLM/BPaL is cost saving, mainly because of reduced time in care and therefore reductions in numbers of outpatient visits, inpatient bed-days and laboratory tests. The panel judged that cost–effectiveness probably favours BPaLM/BPaL.
Equity, acceptability and feasibility
The panel considered the treatment duration and the ability to decentralize treatment (to enable access for remote, underserved settings and disadvantaged populations) to affect equity. Despite not being able to identify relevant research evidence, the panel used their collective experience to judge that there would probably be advantages associated with the use of the BPaLM/BPaL regimen owing to its reduced complexity and shorter duration. Therefore, the panel judged that use of the BPaLM/BPaL regimen would probably increase equity.
A study on the acceptability and feasibility of the BPaL regimen from the provider perspective (29) was considered to be relevant evidence for the assessment of BPaL and indirectly for the assessment of BPaLM. This was a mixed-methods study among a cross section of health care workers, and programmatic and laboratory stakeholders that was carried out between May 2018 and May 2019 in Indonesia, Kyrgyzstan and Nigeria. The results from this study suggested that acceptability and feasibility overall were high. BPaL was rated as acceptable by more than 80% of participants across domains and stakeholders and 88% of interviewed stakeholders stated that they would probably implement BPaL once it became available. Stakeholders appreciated that BPaL would reduce the workload and financial burden on the health care system; expressed concerns about BPaL safety (monitoring), long-term efficacy and national regulatory requirements; and stressed the importance of addressing current health systems constraints, especially in treatment and safety monitoring systems. Results from a second qualitative study (30) with a focus on the patient perspective were presented to the panel; this study suggested that patients would welcome the positive impact of shorter treatment on employment status.20
The panel noted these study results and, as part of their deliberations, they considered patients and health care providers as key stakeholders. The panel considered the following aspects to be critical with regard to the acceptability of BPaLM/BPaL: regimen duration and drug-safety monitoring needs (relating both to the necessary travel, loss of income and general disruption of the life of patients, and to workload for the health care system), and the need for DST. The panel judged that the BPaLM/BPaL regimen would probably be acceptable. Regarding feasibility, the panel noted the limited availability of pure substances of drugs in the BPaLM/BPaL regimen for use in DST as a potential barrier to implementation; they also noted that data on the critical concentration of pretomanid for use in DST are limited. However, given the reduced duration, complexity and associated workload of BPaLM/BPaL, the panel judged that implementation of BPaLM/BPaL is probably feasible.
14 Available at https://clinicaltrials.gov/ct2/home.
15 Trial protocol available at https://trialsjournal.biomedcentral.com/articles/10.1186/s13063-022-06331-8.
16 Several participants excluded in each dataset due to unconfirmed rifampicin resistance.
17 As presented in the expert review (by Dr J-W. Alffenaar, University of Sydney) to the GDG panel in one of the preparatory webinars.
18 Pretomanid has been shown to cause testicular atrophy and impaired fertility in male rats.
19 Estimated regimen prices were calculated using the average weighted price for each medicine (average weighted price accounts for the different prices for each supplier of that medicine weighted by the market share allocation received from each GDF tender), the duration indicated (in months) and assuming 30 days of treatment per month. Actual final costs may differ based on the products delivered.
20 Unpublished, courtesy of Beverley Stringer, Manson unit, Médecins Sans Frontières.