 Feedback
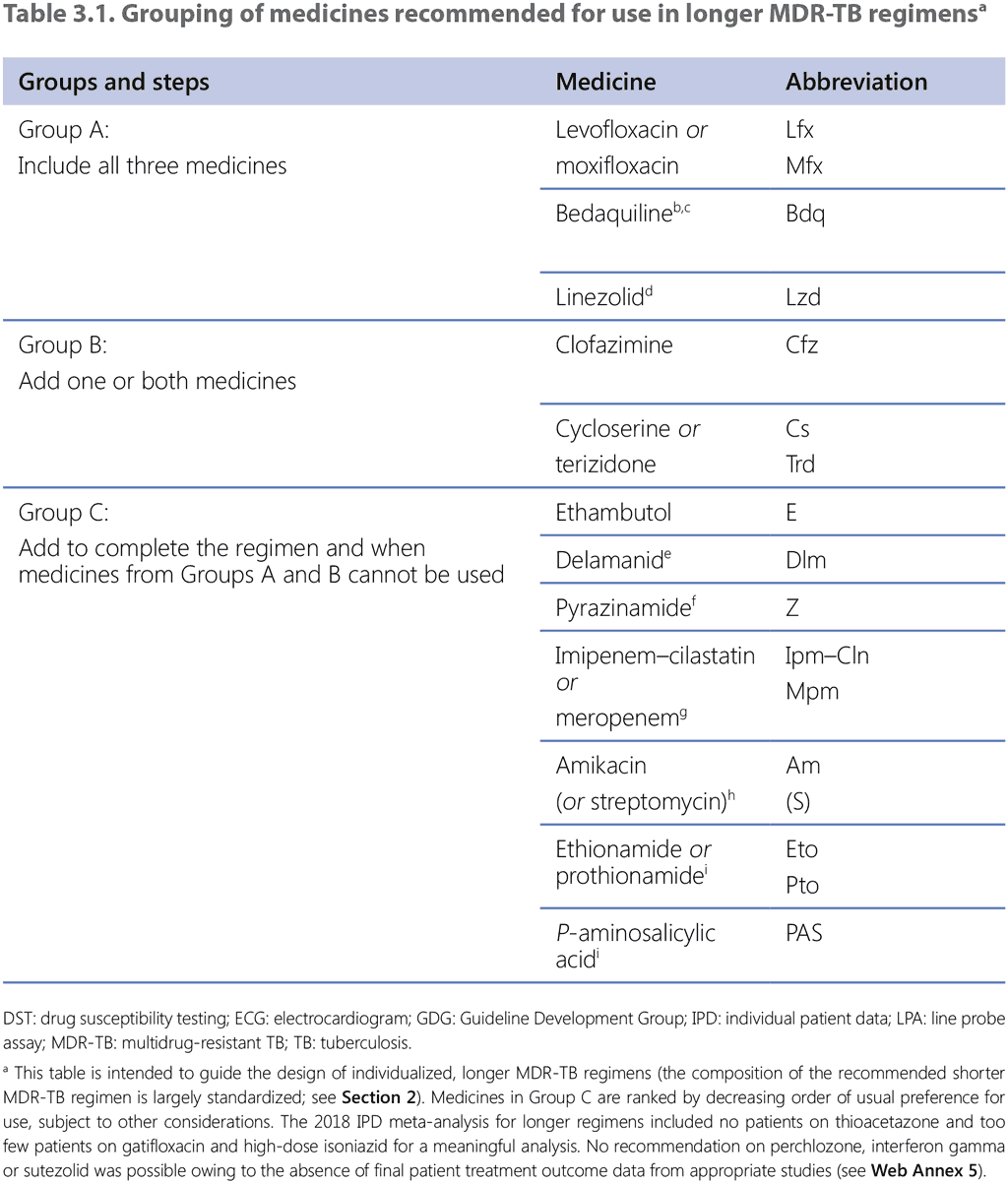
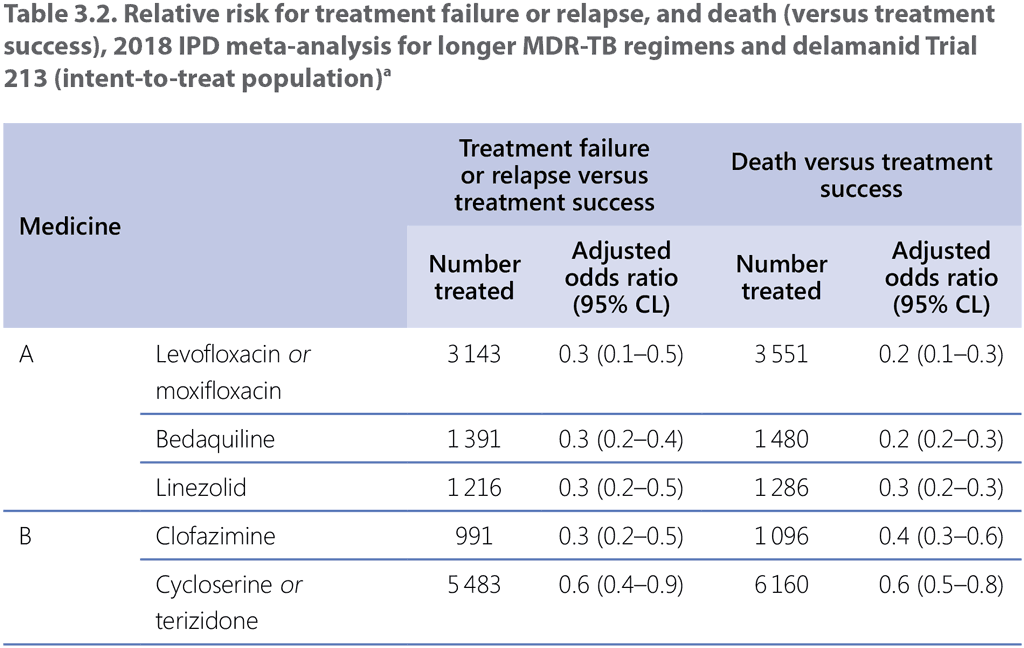
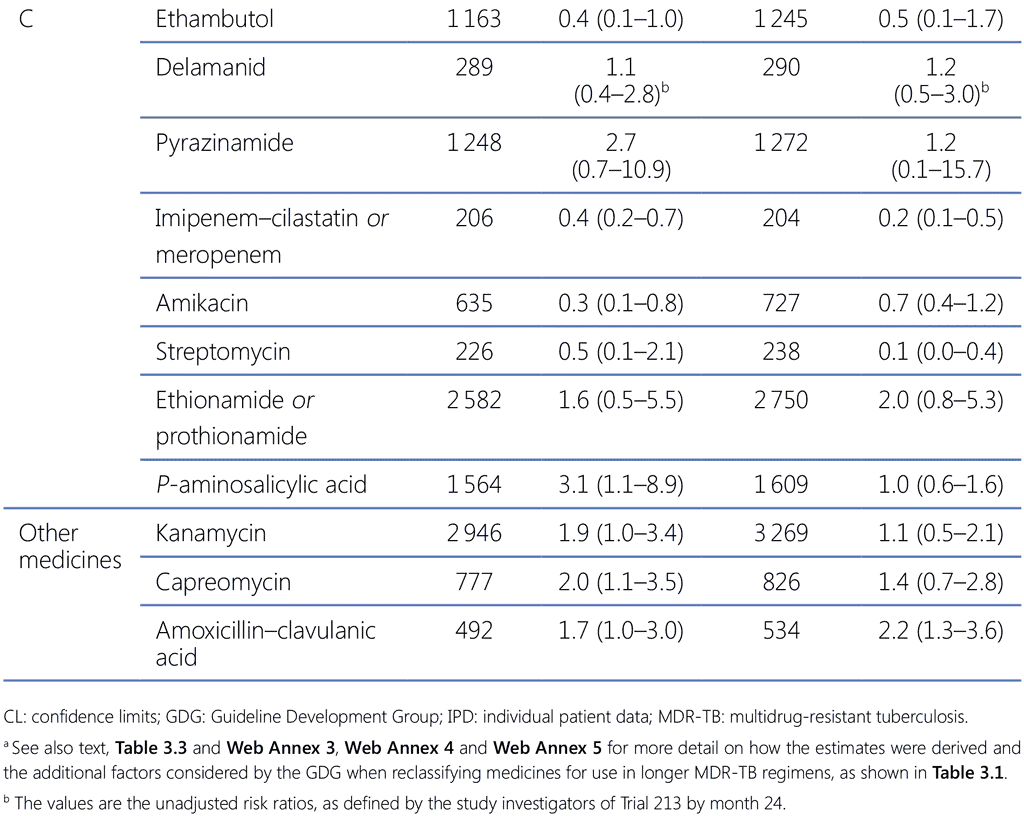
FeedbackThe GDG 2018 assessed the individual contribution to patient outcomes of medicines used in longer MDR-TB regimens, using primarily the estimates of effect from the 2018 IPD meta-analysis and Trial 213 (delamanid) for PICO question 3–2018 (MDR/RR-TB, 2018) (see Web Annex 3 for the respective GRADE summaries of evidence for each medicine, and Web Annex 4 for the evidence-to-decision framework). Following a thorough assessment of the relative benefits and harms, recommendations were made for each medicine and they were classified into three groups (see Table 3.1, Table 3.2 and Table 3.3).
- Group A: fluoroquinolones (levofloxacin and moxifloxacin), bedaquiline and linezolid were considered highly effective and strongly recommended for inclusion in all regimens unless contraindicated.
- Group B: clofazimine and cycloserine or terizidone were conditionally recommended as agents of second choice.
- Group C: included all other medicines that can be used when a regimen cannot be composed with Group A or Group B agents. The medicines in Group C are ranked by the relative balance of benefit to harm usually expected of each.
Other medicines that are not included in Groups A–C are as follows:
Kanamycin and capreomycin – these medicines were associated with poorer outcomes when used; therefore, they are no longer recommended for use in MDR-TB regimens.
Gatifloxacin and high-dose isoniazid, and thioacetazone – gatifloxacin and high-dose isoniazid were used in only a few patients, and thioacetazone was not used at all. Currently, quality-assured preparations of gatifloxacin are not available, following its withdrawal from the market due to concerns about dysglycaemias. Thioacetazone is unlikely to have a role in contemporary longer regimens and is not currently available in a quality-assured formulation. High-dose isoniazid may have a role in patients with confirmed susceptibility to isoniazid (see Section 3.4).
Clavulanic acid – this medicine should be included in MDR/RR-TB regimens only as a companion agent to the carbapenems (imipenem–cilastatin and meropenem). When used in this way, it should be given with every dose of carbapenem, and should not be counted as an additional effective TB agent.
No recommendation on perchlozone, interferon gamma or sutezolid was possible owing to the absence of final patient treatment outcome data from appropriate patient studies.
Regarding the use of bedaquiline in patients aged below 18 years, and considering that exposure– response (efficacy) profiles can be extrapolated from adults to children, the GDG concluded that the doses evaluated in children and adolescents in two trials (Phase 2 trial TMC207-C211 and Phase 1–2 IMPAACT P1108; see Web Annex 5) do not appear to result in exposures that would put patients aged 6–17 years at increased risk for treatment failure. The safety risk in children aged 6 years and older enrolled in the trials – all of whom were HIV-negative and had limited exposure to other QT interval-prolonging medications – did not appear to exceed that of adults. The variability present in the limited sample size precluded a comment on exposure–response (safety). The GDG 2018 also concluded that the risk–benefit considerations for the use of bedaquiline in patients aged 6–17 years are similar to those considered for adults; however, the GDG stressed the need for more data before considering upgrading this recommendation to “strong”.
The GDG review in 2021 determined that the balance between desirable and undesirable effects probably favours the use of bedaquiline in children aged below 6 years. The GDG 2021 highlighted that the benefits may vary depending on specific contexts and population characteristics, such as by nutritional status. The GDG also noted that the potential higher cost of bedaquiline in an MDR/RR-TB treatment regimen should be considered in the context of the benefits of shorter injectable-free regimens (i.e. less travel, reduced time spent in clinics and fewer adverse events). In addition, they judged that equity might increase when bedaquiline becomes available to younger children, because its use would be acceptable to most stakeholders, and that one of the main feasibility aspects would be related to the need for safety monitoring (i.e. access to ECG monitoring, as well as staff capacity for monitoring). However, the panel judged that implementing the use of bedaquiline in young children was probably feasible.
With respect to the use of delamanid in children aged below 6 years, the GDG review in 2018 decided that – based on findings in adults, and on the pharmacological and safety data reviewed – extrapolations on efficacy and safety should be restricted to children aged 3–5 years, but not to children aged below 3 years (see Web Annex 5). Exposure profiles in children aged 3–5 years were comparable to adults, and were no higher than in children aged 6 years and older, for whom past GDGs convened by WHO had already recommended the use of delamanid (10, 64). Based on the laboratory and cardiac data provided, no safety signals distinct from those reported in adults were observed in children aged 3–5 years. The GDG nonetheless had concerns about the feasibility of administering the correct dose to children aged 3–5 years, given that the special formulation used in the trial (25 mg) would not be available in the foreseeable future, and that only the adult tablet (50 mg) is available, which is not bioequivalent and presents challenges to manipulating its contents without compromising its effectiveness.
The GDG review in 2021 concluded that the balance between desirable and undesirable effects probably favours the use of delamanid in children aged below 3 years. The GDG 2021 further stated that when the 25 mg dispersible tablet became available in the future, the resource implications could vary. It was thought that delamanid containing longer treatment regimens could potentially increase equity and be acceptable to stakeholders. In addition, the GDG 2021 judged that it would probably be feasible to use delamanid in children of all ages, especially as the child-friendly formulation of delamanid was expected to become available later in 2021 (this formulation is now available). This judgement also considered that adult tablets cannot be split, crushed or dissolved to ease administration in children without potentially altering bioavailability.
As a result of these multiple reviews as new data have gradually become available, the use of bedaquiline and delamanid are no longer restricted by the age of the patient.

PICO question 4–2018 (MDR/RR-TB, 2018) (number of agents likely to be effective)
Regarding PICO question 4–2018 (MDR/RR-TB, 2018), the analysis showed that in longer MDR-TB treatment regimens, the risk of treatment failure, relapse and death was comparable when the treatment started with four, five or six medicines that were likely to be effective. It also showed that patients who took three agents in the continuation phase – the situation expected when starting with four agents and stopping the injectable agent at the end of the intensive phase – fared no worse than those who took four agents in the continuation phase.
Given that drug–drug interactions, pill burden and likelihood of adverse events all increase with the number of agents in a regimen, it would be desirable to give patients the minimum number of medicines necessary to obtain comparable levels of relapse-free cure. When deciding on the minimum number of agents to recommend, the GDG 2018 considered analyses that included injectable agents in the regimens, while fully cognizant that future longer regimens are expected to be increasingly injectable free. Moreover, it was important to provide for situations in which more than one medicine is stopped at some point during treatment, either because of its indication for use – bedaquiline and delamanid on-label use is 6 months – or because of tolerability (particularly linezolid; Table 3.3) (66); hence, for most of its duration, the regimen would contain two key agents fewer than at the start. Although bedaquiline use beyond 6 months is referred to as off-label use, new evidence on the safety profile of bedaquiline use beyond 6 months was available to the GDG 2019. This evidence supports the safe use of bedaquiline beyond 6 months in patients who receive appropriate schedules of baseline and follow-up monitoring. The use of bedaquiline beyond 6 months continues to be off-label use; thus, best practices in off-label use still apply.
The 2018 IPD included experience from more than 300 patients who were treated with linezolid for at least 1 month, mostly at a dose of 600 mg/day, with information on duration of use. About 30% only received linezolid for 1–6 months, but more than 30% received it for more than 18 months, and these patients had the lowest frequency of treatment failure, loss to follow-up and death. A plot of linezolid duration and treatment failure suggests that the optimal duration of use would be about 20 months, corresponding to the usual total duration of a longer MDR-TB regimen. However, such an analysis does not account for survivorship bias, meaning that those who complete the full length of treatment are more likely to have a successful outcome, given that deaths and losses to follow-up occur earlier. No clear pattern could be discerned for type of adverse event and duration of use, although a few cases were reported with optic neuropathy, known to be associated with long-term use of linezolid (67), whereas haematological toxicity was reported regardless of duration of use.
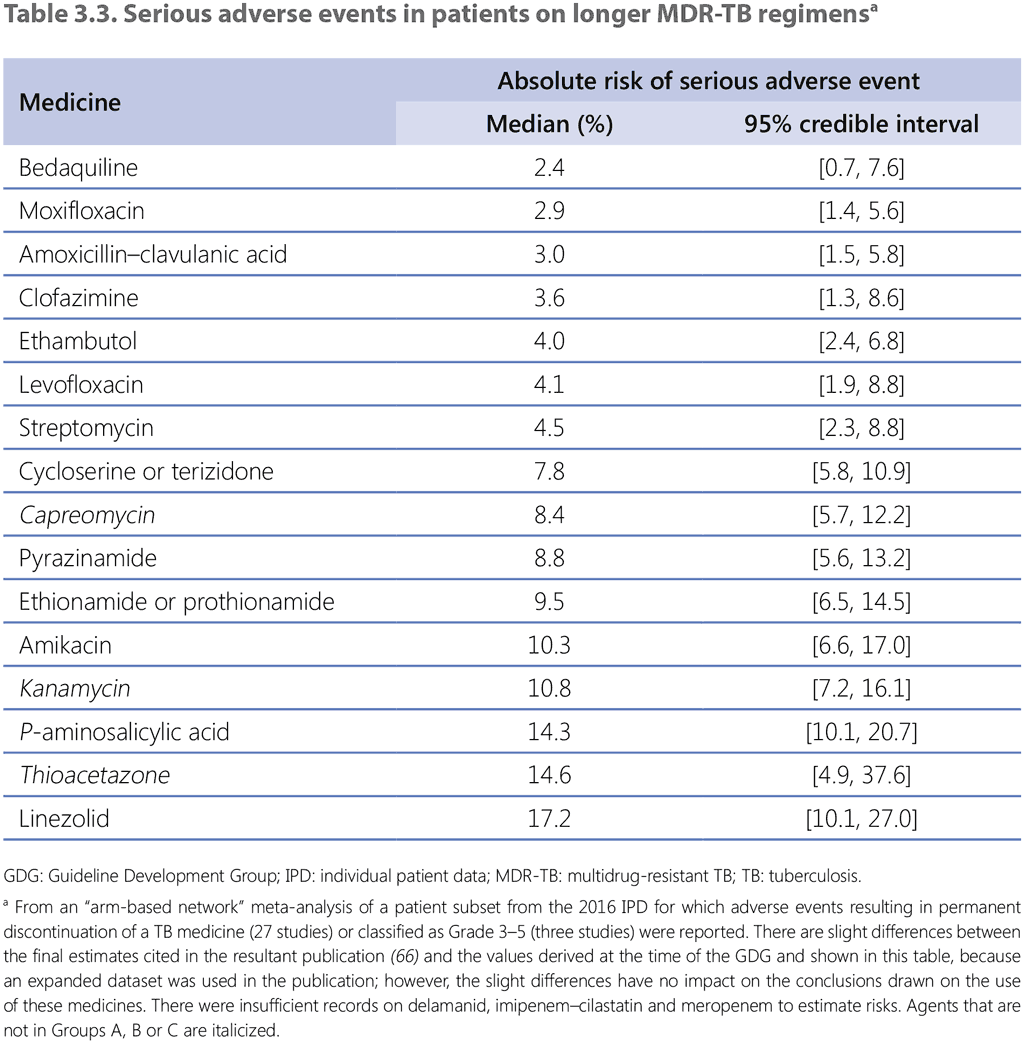
In 2018, the GDG recommended that, where possible, regimens be composed of all three Group A agents and at least one Group B agent, so that treatment starts with at least four medicines likely to be effective, and that at least three agents are continued for the remaining duration of treatment if bedaquiline is stopped after 6 months. New evidence on the safety profile of bedaquiline use beyond 6 months was available to the GDG 2019. This evidence supports the safety of using bedaquiline beyond 6 months in patients who receive appropriate schedules of baseline and follow-up monitoring. If only one or two Group A agents can be used, both Group B agents are included. If the regimen cannot be composed with agents from Groups A and B alone, Group C agents are added to complete it. For patients in whom two agents from Group A are more likely to be stopped before the end of treatment (e.g. pre-existing comorbidities require that both bedaquiline and linezolid be stopped early because of health risks), then starting with five effective agents rather than four may be advisable. These provisions are expected to apply to most MDR/RR-TB patients, including those with additional resistance to fluoroquinolones or other medicines.
PICO question 8–2019 (MDR/RR-TB, 2019) (use of bedaquiline longer than 6 months)
Regarding PICO question 8–2019 (MDR/RR-TB, 2019), the analysis yielded aORs of 1.5 (95% CI: 0.7–2.7) for treatment success versus failure, 0.8 (95% CI: 0.2–0.4) for treatment success versus death, 1.0 (95% CI: 0.5–1.7) for treatment success versus failure or death, and 0.8 (95% CI: 0.5–1.2) for treatment success versus all unfavourable outcomes. The evidence reviewers had planned to use two analytical approaches designed to minimize bias; that is, marginal structural models to account for time-varying confounders, and for exact and propensity score matching of patient characteristics. However, sample size meant that there were limitations in how the first approach could be applied; also, owing to limitations with the dataset, biostatisticians advised that it was not possible to adjust for confounders according to the original data analysis plan. The GDG 2019 noted that the population included in the studies that were assessed was highly selected, with the potential for confounding by indication (i.e. the people who received bedaquiline for >6 months were likely to have done so because of clinical factors that indicated prolonged treatment with bedaquiline). The GDG concluded that there was a high likelihood of residual confounding in the data, and that the patient population addressed in the study did not permit extrapolation to routine use in all MDR/RR-TB patients. This precluded a formal recommendation on the efficacy or effectiveness of bedaquiline use beyond 6 months duration; however, the GDG 2019 concluded that a statement on safety could be made. This information is included in Section 3.5 and in a table note for Table 3.1.
Regarding adverse events, among the 750 patients receiving bedaquiline without concomitant delamanid in the endTB observational study (total exposure of 6 316 person-months), 26 patients experienced a drug-related adverse event (rate: 0.44 per 100 person-months of exposure), with 16 patients having this event classified as a serious adverse event (rate: 0.25 per 100 person-months of exposure). In the first 203 days of exposure to bedaquiline (total exposure of 4 304 person-months), 20 of the 26 drug-related adverse events and 15 of the 16 serious adverse events occurred; the remaining six of the 26 drug-related adverse events and one of the 16 serious adverse events occurred subsequently. All patients who received bedaquiline for more than 203 days did not experience a drug-related adverse event (of any grade) in the first 203 days of treatment. Also, rates of treatment drug-related adverse events appeared to be lower after the first 203 days – at 0.51 in the first 203 days versus 0.30 in the subsequent days per 100 person-months. Similarly, rates of drug-related serious adverse events appeared to be lower after the first 203 days – at 0.35 in the first 203 days versus 0.05 in the subsequent days per 100 person-months.
QTcF values among people receiving bedaquiline increased by an average of 22 ms (from 397 ms to 419 ms) from those taken before or at the time of first receipt of bedaquiline to the end of the first month. In subsequent months of exposure, the mean QTcF values were all lower than at the end of the first month (range: 404–419 ms). Increases in QTcF of more than 60 ms from baseline occurred in about 12% of patients. QTcF prolongation of more than 500 ms was rare, occurring in 0.4–1.5% of patients during each of the first 9 months, but not thereafter. The greatest number of occurrences of QTcF of more than 500 ms happened among people receiving bedaquiline and clofazimine; however, this was also the most common combination of medicines received.
Drug-related cardiac adverse events occurred in 22 people; of these, 15 were among people receiving bedaquiline with clofazimine, but no moxifloxacin or delamanid (rate: 0.3 per 100 person-months), five were among people receiving bedaquiline with clofazimine and moxifloxacin, but no delamanid (rate: 0.3 per 100 person-months), and two were among people receiving bedaquiline and delamanid, regardless of clofazimine and moxifloxacin use (rate: 0.2 per 100 person-months). No events occurred among people receiving bedaquiline without clofazimine, moxifloxacin and delamanid.
Regarding bedaquiline exposure during pregnancy, the findings of the cohort study demonstrated no statistically significant differences in birth or pregnancy outcomes when comparing infants who had intrauterine bedaquiline exposure with those who did not have this exposure (P=0.741 for birth outcomes and P=0.312 for pregnancy outcomes) (51). There were 45 live births (92% of total) in the bedaquiline exposed group compared with 54 live births (90% of total) in the unexposed group. In addition, there were four fetal and neonatal deaths in the infants exposed to bedaquiline (8% of the total bedaquiline exposed group, with three stillbirths and one termination of pregnancy) and six fetal and neonatal deaths in the bedaquiline unexposed group (10% of the total unexposed group, comprising three stillbirths and three miscarriages) (51). The results of the study also demonstrated that treatment outcomes were favourable for pregnant women exposed to bedaquiline compared with those not exposed (71% vs 62%, respectively, P=0.349) (51). Pregnancy outcomes included live births and unfavourable pregnancy outcomes (fetal and neonatal deaths, preterm births <37 weeks and low birth weight <2 500 g); infant outcomes included weight gain and developmental milestones and the diagnosis of TB (51). Of all pregnancy and infant outcomes assessed, only low birth weight was associated with bedaquiline exposure in utero (45% vs 26%, P=0.034). The average weight in bedaquiline exposed infants was 2 690 g versus 2 900 g in infants not exposed to bedaquiline. However, it was not possible to conclusively ascribe this effect to bedaquiline, and more investigation is needed to explore this relationship (51). There were no significant differences in infant growth after birth: in a subanalysis of 86 babies followed up prospectively – 41 exposed to bedaquiline in utero and 45 not exposed – 88% of babies exposed to bedaquiline in utero had normal weight gain at 1 year of age versus 82% of babies not exposed (P=0.914) (51).
PICO question 9–2019 (MDR/RR-TB, 2019) (use of bedaquiline and delamanid together)
Regarding PICO question 9 (MDR/RR-TB, 2019), the analyses yielded aORs of 1.6 (95% CI: 0.5–5.4) for treatment success versus treatment failure, 0.8 (95% CI: 0.3–2.1) for treatment success versus death, 1.2 (95% CI: 0.6–2.5) for treatment success versus failure or death, and 0.6 (95% CI: 0.3–1.1) for treatment success versus all unfavourable outcomes. Regarding adverse events, among the 92 patients receiving bedaquiline with concomitant delamanid during treatment in the endTB observational study (total exposure of 1 095 person-months), two bedaquiline-related adverse events and delamanid-related adverse events occurred (combined rate: 0.46 per 100 person-months of exposure). This rate was comparable to the rates among people receiving bedaquiline alone (0.41 per 100 person-months of exposure) and delamanid alone (0.68 per 100 person-months of exposure). Two drug-related serious adverse events occurred among the 92 patients receiving concomitant bedaquiline and delamanid, one attributed to each drug (combined rate: 0.09 per 100 person-months of exposure). The rate of these events was lower than the rates of drug-related serious adverse events among patients receiving either of these drugs alone (bedaquiline, 0.28; delamanid, 0.39). No fatal drug-related events occurred among patients receiving bedaquiline and delamanid concurrently.
QTcF values among people receiving bedaquiline and delamanid increased by an average of 15 ms (from 398 ms to 413 ms) from those taken before or at the time of first receipt of concurrent bedaquiline and delamanid use, to the end of the first month. In subsequent months of exposure, the mean QTcF values were similar to those at the end of the first month (range: 404–420 ms). QTcF prolongation of more than 500 ms was rare, occurring in only one patient in month 7 of concomitant exposure. Drug-related cardiac adverse events were infrequent, occurring in only two of 92 people exposed to concomitant bedaquiline and delamanid (rate: 0.2 per 100 person-months). Only one drug-related cardiac serious adverse event occurred (rate: 0.1 per 100 person-months). No fatal drug-related cardiac events occurred among the 92 people exposed to bedaquiline and delamanid concurrently.
In the endTB observational study overall (n=1 094), there were two fatal drug-related cardiac events (sudden deaths attributable to QT prolongation), and one other patient experienced a cardiac arrhythmia. The two deaths occurred among patients receiving bedaquiline, clofazimine, capreomycin and p-aminosalicylic acid (but not moxifloxacin or delamanid); in both patients, hypokalaemia was present. These patients were not included in the analysis related to this PICO question because they did not meet the criteria for inclusion according to the predefined statistical analysis plan. However, recognizing that these estimates of serious adverse events were absolute and not relative, the panel felt that this additional evidence was important for close monitoring when the final data of the endTB observational study become available.
The GDG agreed that there was insufficient evidence to assess the efficacy or effectiveness of the concomitant use of bedaquiline and delamanid, given that there were only 84 patients in the intervention group and the data did not lend themselves to a meaningful analysis for the secondary comparator (extended use of delamanid alone) because the populations were too different to allow for the matching that is usually carried out. This precluded a formal recommendation on the efficacy or effectiveness of the concomitant use of bedaquiline and delamanid; however, the GDG concluded that a statement on safety could be made. This information is included in Section 3.5 and in a table note for Table 3.1.
Additional data presented from the DELIBERATE trial highlighted that – among the patients randomized to bedaquiline (n=28), delamanid (n=27) or both medicines (n=27) – the on-treatment change in QTcF from baseline was 11.9 ms, 8.6 ms and 20.7 ms, respectively.34 Of the 27 patients who received both medicines, 10 (37.0%) experienced a Grade 135 QT prolongation adverse event, and two (7.4%) experienced a Grade 2 QT adverse event. In the bedaquiline arm, 32.0% and 3.6% of patients experienced Grade 1 and 2 QT adverse events; in the delamanid arm, these figures were 41.0% for a Grade 1 QT adverse event and 7.4% for a Grade 2 QT adverse event. No patients experienced Grade 3 or 4 QT adverse events. The study investigators concluded that the QTcF prolongation effects of concurrent delamanid and bedaquiline use were not greater than their additive effects. The GDG noted that the QT adverse events in the DELIBERATE trial were surrogate markers of sudden cardiac death. They also noted that levofloxacin was the fluoroquinolone of choice in regimens given to patients in the DELIBERATE trial and that serum potassium was closely monitored.
PICO question 1–2021 (Childhood TB 2021) (use of bedaquiline in MDR/RR-TB patients aged below 6 years) and PICO question 2–2021 (Childhood TB 2021) (use of delamanid in MDR/RR-TB patients aged below 3 years)
Regarding PICO question 1–2021 (Childhood TB 2021) and PICO question 2–2021 (Childhood TB 2021), the details of the evidence review and GDG deliberations can be found in Module 5. Management of tuberculosis in children and adolescents.
34 Personal communication, K Dooley, Johns Hopkins Medicine, November 2019 – for this statement and the rest of this paragraph.
35 In the DELIBERATE trial, a Grade 1 QT adverse event was classified as an absolute QTcF in the following situations: >480 ms and ≤500 ms and QTcF change from baseline from >0 ms to ≤30 ms OR an absolute QTcF ≤480 ms and QTcF change from baseline from >30 ms to ≤60 ms. A Grade 2 QT adverse event was classified as an absolute QTcF in the following situations: >480 ms and ≤500 ms and QTcF change from baseline from >30 ms to ≤60 ms OR an absolute QTcF ≤480 ms and QTcF change from baseline >60 ms. A Grade 3 QT adverse event was classified as an absolute QTcF in the following situation: >500 ms OR an absolute QTcF >480 ms and QTcF change from baseline >60 ms. A Grade 4 QT adverse event was a life-threatening consequence; for example, torsade des pointes or other associated serious ventricular dysrhythmia (personal communication, K Dooley, Johns Hopkins Medicine, November 2019).