 Retour
RetourLa présente section présente les questions PICO posées, les données et les études prises en compte pour répondre aux questions, les méthodes utilisées pour l’analyse et la synthèse des données, un résumé des données sur les effets souhaitables et indésirables et la certitude des données, ainsi qu’un résumé des autres données prises en compte lors de la formulation de la recommandation. D’autres détails sur les données probantes sont disponibles dans les annexes en ligne contenant les tableaux de synthèse des données probantes GRADE (Annex 3) et les tableaux décisionnels GRADE (Annex 4).
Questions PICO
La recommandation de la présente section a été formulée suite à l’évaluation des questions PICO indiquées ci-dessous. En raison des différents groupes d’intervention et de comparaison utilisés, les questions PICO 3, 5 et 6 ont été divisées en plusieurs sous-questions PICO (les détails figurent dans le texte et dans le Tableau 1.3).
Question PICO 3–2022 (TB MR-RR, 2022) : Faut-il utiliser les schémas BPaL avec une exposition plus faible au linézolide (dose ou durée) plutôt que le schéma BPaL original chez les patients éligibles au schéma BPaL ?
Question PICO 4–2022 (TB MR-RR, 2022) : Faut-il utiliser un schéma thérapeutique de 6 mois à base de bédaquiline, de prétomanide et de linézolide chez les patients atteints de tuberculose pulmonaire pré-UR (tuberculose MR-RR-avec résistance aux fluoroquinolones) ?
Question PICO 5–2022 (TB MR-RR, 2022) : Faut-il utiliser un schéma thérapeutique de 6 mois à base de bédaquiline, prétomanide et linézolide chez les patients atteints de tuberculose MR-RR pulmonaire sans résistance aux fluoroquinolones ?
Question PICO 6–2022 (TB MR-RR, 2022) : Faut-il utiliser un schéma thérapeutique de 6 mois à base de bédaquiline, prétomanide et linézolide avec ou sans ajout de moxifloxacine (BPaLM) ou de clofazimine chez les patients atteints de tuberculose pulmonaire MR-RR (avec ou sans résistance aux fluoroquinolones) ?
Données et études prises en compte
L’examen de ces questions PICO lors de la réunion du GDG organisée par l’OMS en février-mars 2022 reposait sur de nouvelles données fournies par MSF dans le cadre de l’essai clinique TB-PRACTECAL et par l’Alliance mondiale pour la mise au point de médicaments antituberculeux dans le cadre de l’essai ZeNix. Pour plusieurs évaluations de ces questions PICO, les données issues de l’ensemble de données individuelles des patients (DIP) de l’OMS de 2021 ont été utilisées. Les populations de patients incluses dans deux essais ont été recrutées selon des critères stricts d’inclusion et d’exclusion ; elles présentaient de nombreuses similitudes et peu de différences notables. Les principaux critères utilisés dans ces essais figurent dans le Tableau 1.1. Pour une liste complète des critères d’exclusion, voir l’ Annexe 2 et les protocoles d’essai publiés.¹⁴
Tableau 1.1. Résumé des principaux critères d’inclusion et d’exclusion : essais TB-PRACTECAL et ZeNix, listes complètes en Annexe 2

Bdq : bédaquiline ; IMC : indice de masse corporelle ; Dlm : délamanide ; ECG : électrocardiogramme ; VIH : virus de l’immunodéficience humaine ; Lzd : linézolide ; MAO : monoamine oxydase ; TB MR-RR : tuberculose multirésistante ou résistante à la rifampicine ; P : rifapentine ; QTcF : intervalle QT corrigé par Fredericia ; TB RR : tuberculose résistante à la rifampicine ; TB : tuberculose ; tdp : torsades de pointes ; TB-UR : tuberculose ultrarésistante.
TB-PRACTECAL
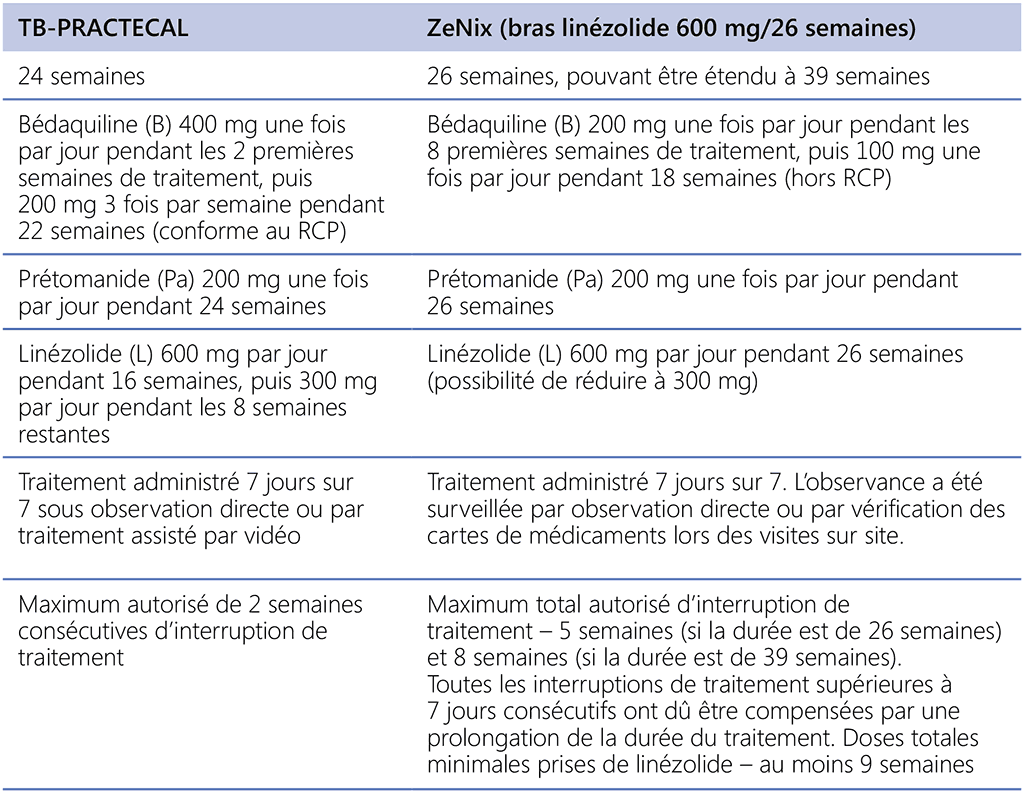
TB-PRACTECAL était un essai de phase 2-3 multicentrique, ouvert, multibras, randomisé, contrôlé, multi-étapes portant sur les schémas thérapeutiques courts contenant de la bédaquiline et du prétomanide en association avec des médicaments antituberculeux existants et repositionnés (p. ex. linézolide et clofazimine) pour le traitement de la tuberculose pulmonaire MR-RR confirmée microbiologiquement.¹⁵
L’étude était divisée en deux stades, avec une transition fluide entre les stades, ce qui signifie que le recrutement dans un bras prenait fin uniquement après qu’une décision ait été prise suite à l’analyse des données du critère d’évaluation principal du stade 1. Dans le premier stade – équivalent à un essai de phase 2B sur l’innocuité et l’efficacité préliminaire – les patients ont reçu de façon aléatoire l’un des quatre schémas, stratifiés par site. Les schémas expérimentaux comprenaient la bédaquiline, le prétomanide et le linézolide par voie orale. Deux des schémas comprenaient également la moxifloxacine (bras 1) et la clofazimine (bras 2). L’objectif principal du stade 1 était de sélectionner les schémas pour l’évaluation au stade 2, sur la base de critères d’évaluation de l’innocuité et de l’efficacité à 8 semaines. Les bras expérimentaux qui ne répondaient pas aux critères prédéfinis d’innocuité et d’efficacité n’étaient pas pris en compte pour une évaluation plus approfondie.
Le deuxième stade de l’étude correspondait à un essai de phase 3 portant sur l’innocuité et l’efficacité du schéma le plus prometteur. Comme prévu dans le protocole de l’étude, l’innocuité et l’efficacité du schéma thérapeutique ont été évaluées par rapport au bras de référence 72 semaines après la randomisation. Le stade 2 de l’essai comprenait un bras d’intervention de BPaLM comparé au traitement de référence approuvé localement, conforme aux recommandations de l’OMS pour le traitement de la tuberculose MR-RR ou de la tuberculose pré-UR au moment de la conduite de l’essai (comprenant un schéma thérapeutique injectable de 9 à 12 mois ; un schéma de 18–24-mois recommandé par l’OMS [avant 2019] ; un schéma entièrement oral de 9 à 12 mois ; et un schéma entièrement oral de 18 à 20 mois). L’essai TB-PRACTECAL a cessé de recruter des patients peu de temps après que son comité indépendant de surveillance de la sécurité des données a indiqué que le schéma BPaLM était plus efficace que le traitement de référence, car il a été considéré peu probable que des données supplémentaires modifient les résultats de l’essai. Cet essai n’a pas été conçu pour comparer les schémas thérapeutiques expérimentaux entre eux.
Les patients éligibles étaient âgés de 15 ans et plus, étaient atteints d’une tuberculose confirmée bactériologiquement (tests moléculaires ou phénotypiques) et présentaient une résistance au moins à la rifampicine établie par un test de sensibilité aux médicaments moléculaire ou phénotypique. Le principal critère d’efficacité était le critère composite d’issues défavorables (échec, décès, arrêt du traitement, récidive ou patients perdus de vue) 72 semaines après la randomisation. Les critères d’efficacité secondaires pertinents étaient notamment la conversion de culture à 12 et 24 semaines, les issues défavorables à 24 semaines après la randomisation, les issues défavorables 108 semaines après la randomisation, le délai médian de conversion de culture et la récidive à la semaine 48 dans les bras expérimentaux. Les participants étaient randomisés selon un ratio de 1:1:1:1 dans le bras de traitement de référence ou l’un des trois bras d’intervention suivants :
- Bras 1 : 24 semaines de B-Pa-Lzd-Mfx (BPaLM) ;
- Bras 2 : 24 semaines de B-Pa-Lzd-Cfz (BPaLC) ; et
- Bras 3 : 24 semaines de B-Pa-Lzd (BPaL).
Dans tous les bras d’intervention, le linézolide a été administré à une dose de 600 mg par jour pendant 16 semaines, puis 300 mg par jour pendant les 8 semaines restantes (ou plus tôt lorsqu’il était modérément toléré). La bédaquiline a été administrée à une dose de 400 mg une fois par jour pendant 2 semaines, puis 200 mg trois fois par semaine pendant 22 semaines. La surveillance de l’innocuité pour la plupart des participants comprenait plusieurs électrocardiogrammes (ECG) au départ, puis une fois par semaine jusqu’à la semaine 8, toutes les 4 semaines jusqu’à la semaine 24, puis toutes les 8 semaines. La surveillance microbiologique comprenait l’examen microscopique de frottis et la culture au départ et au jour 7, puis toutes les 4 semaines jusqu’à la semaine 24 et toutes les 8 semaines par la suite.
ZeNix
ZeNix était un essai randomisé de phase 3 à insu partiel pour évaluer l’innocuité et l’efficacité de diverses doses et durées de traitement du linézolide plus bédaquiline et prétomanide chez les personnes atteintes de tuberculose pulmonaire MR-RR avec une résistance supplémentaire aux fluoroquinolones (avec ou sans résistance aux produits injectables) ou chez les personnes atteintes de tuberculose MR-RR intolérantes au traitement ou ne répondant pas au traitement. Les patients éligibles étaient âgés de 14 ans et plus, pesaient au moins 35 kg, disposaient d’un résultat attesté de test VIH et avaient une tuberculose UR à culture positive confirmée bactériologiquement (définition avant 2021) ou une tuberculose MR-RR confirmée bactériologiquement, mais étaient intolérants au traitement ou ne répondaient pas au précédent traitement contre la tuberculose MR-RR. Le principal critère de jugement était l’incidence de l’échec bactériologique, de la rechute ou de l’échec clinique par un suivi jusqu’à 26 semaines après la fin du traitement. Le critère de jugement secondaire était l’incidence de l’échec bactériologique, de la rechute ou de l’échec clinique par un suivi jusqu’à 78 semaines après la fin du traitement. Les participants ont reçu 26 semaines de traitement avec BPaL. La dose et la durée du linézolide variaient pour chacun des quatre bras : 1 200 mg 26 semaines, 1 200 mg 9 semaines, 600 mg 26 semaines ou 600 mg 9 semaines. La bédaquiline a été administrée à une dose de 200 mg une fois par jour pendant 8 semaines, puis 100 mg une fois par jour pendant 18 semaines. Ce schéma posologique hors RCP est corroboré par des simulations pharmacocinétiques pour un autre schéma posologique de bédaquiline qui donne des expositions comparables et a été élaboré pour favoriser l’observance du traitement et en faciliter l’administration (tous les médicaments chaque jour tout au long du schéma) (23).
La surveillance de l’innocuité comprenait des tests et des évaluations programmés des paramètres biologiques, de l’ECG, des signes vitaux ainsi que d’autres examens cliniques (24). La surveillance microbiologique comprenait l’examen microscopique de frottis, les tests moléculaires et la culture liquide à partir d’expectorations au départ puis à chaque visite des patients (24).
Tableau 1.2. Posologie, administration du traitement et tolérances des modifications du traitement liées à la toxicité

DIP de l’OMS 2021
En 2021, l’OMS a lancé un appel public à la transmission de données permettant de constituer un groupe témoin (traitement de référence) auquel les schémas posologiques de 6 à 9 mois pourraient être comparés. Ces cohortes recevaient un traitement conforme aux lignes directrices de l’OMS sur la tuberculose pharmacodépendante de 2020 avec de la bédaquiline et du linézolide pour une durée allant de 6 à 24 mois. Les patients recevant des antibiotiques injectables étaient exclus.
Les ensembles de données inclus comprenaient des personnes utilisant l’un des schémas suivants :
- schémas thérapeutiques entièrement oraux de 6 à 12 mois utilisant au moins la bédaquiline et le linézolide ; ou
- schéma de l’OMS (2019) entièrement oral sur 9 à 12 mois contenant de la bédaquiline dans l’association, tel que 4-6 Bdq (6 m)-Lfx / Mfx-Cfz-Z-E-Hh-Eto / 5 m Lfx / Mfx-Cfz-Z-E ; ou
- schéma thérapeutique entièrement oral ≥ 18 mois de l’OMS (2018) contenant au moins de la bédaquiline et du linézolide.
Les ensembles de données individuels inclus dans cette cohorte sont décrits en détail dans le plan d’analyse statistique (Annex 6). Pour pouvoir être admis dans un schéma de comparaison court (cible de 9 à 12 mois au début du traitement), les patients doivent avoir rempli chacun des critères suivants :
- avoir eu un traitement n’excédant pas une durée de 12 mois ;
- avoir reçu six médicaments ou plus pendant le traitement, dont la bédaquiline ; et
- en cas de guérison ou de traitement complété, avoir eu un traitement de 8,5 mois ou plus.
Pour être admis dans un schéma de comparaison plus long (cible de 18 à 24 mois), les patients doivent avoir rempli chacun des critères suivants :
- être classés dans l’ensemble de données comme ayant reçu un schéma plus long (si indiqué) ;
- avoir eu un traitement sur une durée n’excédant pas 24 mois ;
- avoir reçu quatre médicaments ou plus (quelle que soit la sensibilité aux médicaments ; c’est-à-dire indépendamment du fait qu’ils soient susceptibles d’être efficaces), y compris la bédaquiline ; et
- en cas de guérison ou de traitement complété, avoir eu un traitement sur une durée minimale de 17,5 mois.
Méthodes utilisées pour l’analyse et la synthèse des données
Des analyses descriptives des caractéristiques de départ des participants dans toutes les études incluses ont été effectuées ; les caractéristiques incluaient les données démographiques, les résultats des tests diagnostiques, les schémas thérapeutiques et les résultats thérapeutiques
Des analyses comparatives ont été réalisées au sein d’études individuelles et entre plusieurs études :
- Comparaisons au sein des études – pour les études dans lesquelles à la fois un schéma court (6 mois) et un comparateur pertinent sont utilisés, des comparaisons par paires étaient effectuées entre chacun des schémas courts et le comparateur. Pour les ECR inclus (p. ex. les essais TB-PRACTECAL et NExT), le résultat principal de l’analyse prédéfinie a également été calculé et communiqué.
- Comparaisons par paires entre les études – des comparaisons portant sur chacune des questions PICO ont été effectuées en comparant les résultats entre les cohortes dans lesquelles les participants ont reçu soit le schéma d’intervention soit le schéma témoin relatif à cette question.
Modèles statistiques
Pour des comparaisons entre les ensembles de données ou les cohortes, les résultats ont été présentés comme des rapports de risques (RR) ajustés et non ajustés. Les rapports de risques ajustés (RRa) ont été calculés en utilisant une régression linéaire généralisée log-binomiale (distribution d’erreur binomiale avec fonction lien logit). Les facteurs de confusion potentiels prédéfinis ont été ajustés pour utiliser la pondération par l’inverse de la probabilité en utilisant le score de propension. Aucun problème de convergence n’est apparu avec le modèle log-binomial. Lorsque les taux de résultats étaient proches de la limite, les RRa n’étaient pas calculés, et les RR non ajustés étaient présentés. Pour les résultats où le nombre d’événements est nul, une différence de risques (DR) non ajustée a été calculée. Pour les DR ou RR non ajustés, des intervalles de confiance (IC) à 95 % ont été calculés en utilisant la méthode des scores. La sélection des covariables pour le calcul des scores de propension était basée sur la disponibilité des données et les connaissances cliniques. Les covariables prises en compte pour l’inclusion dans l’analyse des scores de propension comprenaient l’âge, le genre, le résultat de frottis au départ, le statut par rapport au VIH (y compris le statut par rapport au TARV), les antécédents de traitement (y compris si l’infection antérieure était résistante aux médicaments), l’indice de masse corporelle (IMC), le statut tabagique, le diagnostic de diabète, la cavitation au départ, la présence d’une maladie bilatérale et la résistance à la fluroquinolone. Pour le calcul des RRa, l’imputation multiple par équations de chaîne en utilisant l’approche « dans » le score de propension a été utilisée pour tenir compte des données manquantes dans les facteurs de confusion potentiels lorsque la proportion de valeurs manquantes pour un facteur de confusion était inférieure à 45 %.
Calendrier de suivi pour les comparaisons entre les schémas
Les analyses effectuées pour cet examen des données ont combiné les résultats des cohortes avec différents calendriers de suivi après le début du traitement. Il y a eu des différences dans le calendrier de suivi entre les cohortes (de 5,5 mois à 24 mois) et au sein des cohortes individuelles (par exemple, l’ensemble de DIP 2021 de l’OMS combinait plusieurs cohortes avec des calendriers de suivi variables). Le calendrier de suivi a été divisé entre le temps écoulé entre le début et la fin du traitement d’une part et la période entre la fin du traitement et la fin du suivi d’autre part. Pour les schémas plus courts, le suivi post-traitement était particulièrement important, car des taux de rechute plus élevés peuvent être la conséquence de traitements plus courts qui n’éliminent pas totalement M. tuberculosis. Dans la mesure du possible, il était important que la durée de suivi entre deux groupes dans une comparaison soit équivalente, de sorte que la probabilité de décès ou de rechute soit équivalente pour les participants. Dans ces analyses, la durée de suivi a été mesurée à partir de la date de début du traitement plutôt qu’après la date de fin du traitement, afin de minimiser l’effet des différences dans le temps de suivi.
Les principes de comptabilisation des périodes de suivi étaient les suivants :
- Dans la mesure du possible, suivre les participants dans les groupes d’intervention et les groupes témoins pendant la même durée totale, de sorte que la probabilité d’issues défavorables (p. ex. décès) soit la même dans les deux groupes.
- Limiter le suivi à 24 mois après le début du traitement pour toutes les cohortes. Il n’y a pas eu d’analyse dans laquelle les cohortes des groupes d’intervention et de comparaison faisaient l’objet d’un suivi de plus de 24 mois. Les données recueillies à partir des essais sur le traitement antituberculeux démontrent qu’une proportion élevée de récidives peut survenir dans les 12, voire 6 mois suivant l’arrêt du traitement (25).
- Sélectionner une analyse primaire qui optimise le nombre de participants inclus dans les deux groupes. Pour les schémas plus courts (de 6 à 9 mois), la durée du suivi dans la comparaison a été incluse pour permettre de mettre en évidence les rechutes.
Lorsque c’était possible, des analyses de sensibilité supplémentaires ont été réalisées pour évaluer l’effet de la durée du suivi sur les résultats thérapeutiques.
Résumé des données sur les effets souhaitables et indésirables et certitude quant aux données
Les données relatives aux nouveaux schémas thérapeutiques sur lesquelles reposent les questions du PICO provenaient de deux essais. Il s’agissait d’informations sur un total de 419 des 423 participants qui étaient recrutés dans quatre bras de l’étude TB-PRACTECAL et sur 172 des 181 participants qui étaient recrutés dans quatre bras de l’étude ZeNix¹⁶.
Les données des patients des bras concernés ont été utilisées dans chacune des comparaisons qui ont conduit aux conclusions et à la recommandation finale sur l’utilisation du schéma BPaLM/BPaL. Même si l’essai TB-PRACTECAL n’a pas été conçu pour comparer les schémas thérapeutiques expérimentaux entre eux et avec les traitements de référence, les différents bras de l’essai ont été comparés au bras BPaLM (sous-questions PICO de 6.2 à 6.6) pour aider le groupe à prendre des décisions finales.
Sous-question PICO 3.2
Le bras BPaL 1200–9 de l’essai ZeNix (linézolide 1 200 mg par jour pendant 9 semaines) a été comparé au bras BPaL 1200–26 (linézolide 1 200 mg par jour pendant 26 semaines) dans la même population de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones. L’analyse primaire a été effectuée 12 mois après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaL avec du linézolide 1200–9 (n=43) comparés aux participants avec les mêmes schémas de résistance recevant le BPaL avec du linézolide 1200–26 (n=44) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique inférieurs (93 % contre 98 %) ; soit une réduction relative de 5 % (RR=0,95, IC 95 % : 0,87 à 1,05) ;
- des taux d’échec et de rechute supérieurs (4,7 % contre 2,3 %) ; soit une augmentation relative par deux (RR=2,1, IC 95 % : 0,19 à 22) ;
- des taux de décès supérieurs (2,3 % contre 0 %) ; soit une augmentation absolue de 2 % (DR=0,02, IC 95 % : –0,06 à 0,12) ;
- des taux de perdus de vue identiques (0 % contre 0 %) ; soit une différence absolue de 0 % (DR=0,00, IC 95 % : –0,08 à 0,08) ;
- des taux d’événements indésirables inférieurs (16 % contre 18 %) ; soit une réduction relative de 10 % (RR=0,90, IC 95 %: 0,36 à 2,3) ; et
- des taux d’amplification de la pharmacorésistance identiques (0 % contre 0 %) ; soit une différence absolue de 0 % (DR = 0,00, IC 95 % : –0,08 à 0,08).
Le GDG a estimé que les effets bénéfiques du BPaL avec le linézolide 1200–9 étaient faibles et que les effets indésirables étaient modérés par rapport au BPaL avec le linézolide 1200–26. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du BPaL avec le linézolide 1200–26.
Conclusion
L’utilisation du linézolide 1 200 mg pendant 26 semaines, plutôt que du linézolide 1 200 mg pendant 9 semaines, est préconisée dans le cadre du schéma BPaL chez les adultes atteints de tuberculose MR-RR ou de tuberculose pré-UR.
Sous-question PICO 3.3
Le bras BPaL 600–26 de l’essai ZeNix (linézolide 600 mg par jour pendant 26 semaines) a été comparé au bras BPaL 1200–26 (linézolide 1 200 mg par jour pendant 26 semaines) dans la même population de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones. L’analyse primaire a été effectuée 12 mois après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaL avec du linézolide 600–26 (n=43) comparés aux participants avec les mêmes schémas de résistance recevant le BPaL avec du linézolide 1200–26 (n=44) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (100 % contre 98 %) ; soit une augmentation relative de 2 % (RR=1,02, IC 95 % : 0,98 à 1,07) ;
- des taux d’échec et de rechute inférieurs (0 % contre 2,3 %) ; soit une réduction absolue de 2 % (DR= –0.02, IC 95 % : –0,12 à 0,06) ;
- des taux d’événements indésirables de grade 3–5 inférieurs (14 % contre 18,6 %) ; soit une réduction relative de 23 % (RR=0,77, IC 95 %: 0,29 à 2,03) ; et
- des taux de décès (0 % contre 0 %), de perdus de vue (0 % contre 0 %) ou de résistance amplifiée (0 % contre 0 %) identiques.
Le GDG a estimé que les effets bénéfiques du BPaL avec le linézolide 600-26 étaient modérés et que les effets indésirables étaient négligeables par rapport au BPaL avec le linézolide 1200-26. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du BPaL avec le linézolide 600–26.
Conclusion
L’utilisation de linézolide 600 mg pendant 26 semaines, plutôt que du linézolide 1200 mg pendant 26 semaines, est préconisée dans le cadre du schéma BPaL chez les adultes atteints de tuberculose MR-RR ou de tuberculose pré-UR.
Sous-question PICO 3.4
Le bras BPaL 600-9 de l’essai ZeNix (linézolide 600 mg par jour pendant 9 semaines) a été comparé au bras BPaL 1200-26 (linézolide 1 200 mg par jour pendant 26 semaines) dans la même population de patients atteints de tuberculose MR-RR-avec ou sans résistance aux fluoroquinolones. L’analyse primaire a été effectuée 12 mois après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le BPaL avec le linézolide 600-9 (n = 42) comparés aux participants avec les mêmes schémas de résistance recevant le BPaL avec le linézolide 1200-26 (n = 44) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique inférieurs (93 % contre 98 %) ; soit une réduction relative de 5 % (RR=0,95, IC 95 %: 0,86 à 1,05) ;
- des taux d’échec et de rechute supérieurs (4,8 % contre 2,3 %) ; soit une augmentation relative par deux (RR=2,10, IC 95 % : 0,20 à 22,26) ;
- des taux de perdus de vue supérieurs (2,4 % contre 0 %) ; soit une augmentation absolue de 2 % (DR=0,02, IC 95 % : –0,06 à 0,12) ;
- des taux d’événements indésirables de grade 3 à 5 inférieurs (14,3 % contre 18,2 %) ; soit une réduction relative de 21 % (RR=0,79, IC 95 %: 0,30 à 2,07) ; et
- des taux de décès (0 % contre 0 %) ou de résistance amplifiée (0 % contre 0 %) identiques.
Le GDG a estimé que les effets bénéfiques du BPaL avec le linézolide 600–9 étaient faibles et que les effets indésirables étaient modérés par rapport au BPaL avec le linézolide 1200–26. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du BPaL avec le linézolide 1200–26.
Conclusion
L’utilisation de linézolide 1200 mg pendant 26 semaines, plutôt que du linézolide 600 mg pendant 9 semaines, est préconisée dans le cadre du schéma BPaL chez les adultes atteints de tuberculose MR-RR ou de tuberculose pré-UR.
Question PICO 3 – Conclusion récapitulative intermédiaire
L’évaluation de la question PICO 3 a permis de prendre une décision sur la dose et la durée optimales du linézolide dans le schéma BPaLM/BPaL, et de limiter les comparaisons ultérieures au schéma d’intervention avec cette dose et cette durée particulières de linézolide – BPaL (600 mg – 26 semaines).
Sous-question PICO 4.1
Le bras BPaL 600–26 de l’essai ZeNix (dans lequel le linézolide 600 mg par jour a été utilisé pendant 26 semaines et la population comprenait des patients atteints de tuberculose MR-RR avec résistance aux fluoroquinolones) a été comparé à une cohorte de patients atteints de tuberculose MR-RR avec résistance aux fluoroquinolones des DIP de 2021 qui recevaient des traitements plus longs pour le traitement de la tuberculose MR-RR, conçus conformément aux lignes directrices 2020 de l’OMS. L’analyse primaire a été effectuée 18 mois après le début du traitement.
Les participants atteints de tuberculose pulmonaire pré-UR (tuberculose MR-RR avec résistance aux fluoroquinolones) recevant le BPaL 600–26 (n=33) comparés aux participants recevant des traitements plus longs pour la tuberculose MR-RR (n=839) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (100 % contre 75 %) ; soit une augmentation relative de 34 % (RR=1,34, IC 95 % : 1,20 à 1,40) ;
- des taux de succès thérapeutique supérieurs (100 % contre 75 %) ; soit une augmentation relative
de 34 % (RR=1,34, IC 95 % : 1,20 à 1,40) ; - des taux de décès inférieurs (0 % contre 9,9 %) ; soit une réduction absolue de 10 % (DR= –0.10, IC 95 % : –0.12 à –0,01) ;
- des taux de perdus de vue inférieurs (0 % contre 9,1 %) ; soit une réduction absolue de 9 % (DR=–0,09, IC 95 % : –0.11 à –0.01) ; des taux d’événements indésirables plus élevés (15 % contre 4,4 %) ; soit une augmentation de 3,4 (RR=3,44, IC 95 % : 1,44 à 8,17) ; et
- des taux d’amplification de la pharmacorésistance inférieurs (0 % contre 7,4 %) ; soit une réduction absolue de 7 % (DR= –0,07, IC 95 % : –0.09 à –0.03).
Le GDG a estimé que les effets bénéfiques du BPaL avec le linézolide 600-26 étaient importants et que les effets indésirables étaient modérés par rapport aux schémas plus longs recommandés par l’OMS. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du BPaL avec le linézolide 600–26.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide et de linézolide (BPaL), plutôt que d’un schéma plus long (18 mois), est préconisée chez les patients atteints de tuberculose MR-RR avec une résistance aux fluoroquinolones (tuberculose pré-UR), qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins de 1 mois.
Question PICO 4 – Conclusion intermédiaire
L’évaluation de la question PICO 4 a conduit à la recommandation conditionnelle pour l’utilisation du schéma BPaL (600 mg – 26 semaines) plutôt que des schémas plus longs actuellement recommandés chez les patients atteints de tuberculose MR-RR avec une résistance supplémentaire aux fluoroquinolones (tuberculose pré-UR).
Sous-question PICO 5.1
Le bras BPaL 600–26 de l’essai ZeNix (dans lequel le linézolide 600 mg par jour a été utilisé pendant 26 semaines et la population comprenait des patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones) a été comparé à une cohorte de patients atteints de tuberculose MR-RR sans résistance aux fluoroquinolones traités en Afrique du Sud avec le schéma sur 9 mois recommandé par l’OMS par éthionamide pendant 4 mois. L’analyse primaire a été effectuée 12 mois après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaL 600–26 (n=43) comparés aux participants atteints de tuberculose MR-RR (sans résistance aux fluoroquinolones) recevant le schéma de 9 mois avec l’éthionamide (n=785) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (100 % contre 69 %) ; soit une augmentation relative de 45 % (RR=1,45, IC 95 % : 1,32 à 1,53) ;
- des taux d’échec et de rechute inférieurs (0 % contre 1,3 %) ; soit une réduction absolue de 1 % (DR=–0,01, IC 95 % : –0,02 à 0,07) ;
- des taux de décès inférieurs (0 % contre 19 %) ; soit une réduction absolue de 19 % (DR=–0,19, IC 95 % : –0,22 à –0,1) ;
- des taux de perdus de vue inférieurs (0 % contre 11 %) ; soit une réduction absolue de 11 % (DR = –0.11, IC 95 % : –0,14 à –0,03) ; et
- des taux de résistance amplifiée identiques (0 % contre 0 %) ; soit une différence absolue de 0 % (DR = 0,00, IC 95 % : –0,01 à 0,08).
Des événements indésirables de grade 3 à 5 ont été observés chez 14 % des participants recevant le BPaL 600–26, mais aucune comparaison n’a pu être effectuée, car aucune donnée n’était disponible pour les participants recevant le traitement de 9 mois par éthionamide.
Le GDG a estimé que les effets bénéfiques du BPaL avec le linézolide 600-26 étaient importants et que les effets indésirables étaient modérés par rapport au schéma de 9 mois avec l’éthionamide recommandé par l’OMS. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du BPaL avec le linézolide 600–26.
Conclusion
L’utilisation d’un schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide et de linézolide (BPaL), plutôt que d’un schéma de 9 mois (avec de l’éthionamide), est préconisée chez les patients atteints de tuberculose MR-RR sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins de 1 mois.
Sous-question PICO 5.2
Le bras BPaL 600–26 de l’essai ZeNix (dans lequel le linézolide 600 mg par jour a été utilisé pendant 26 semaines et la population comprenait des patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones) a été comparé à une cohorte de patients atteints de tuberculose MR-RR sans résistance aux fluoroquinolones des DIP de 2021, recevant des schémas plus longs pour le traitement de la tuberculose MR-RR, conçus conformément aux lignes directrices 2020 de l’OMS. L’analyse primaire a été effectuée 18 mois après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaL 600–26 (n=43) comparés aux participants atteints de tuberculose MR-RR (sans résistance aux fluoroquinolones) recevant des schémas plus longs recommandés par l’OMS (n = 850) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (98 % contre 74 %) ; soit une augmentation relative de 32 % (RR=1,32, IC 95 %: 1,19 à 1,39) ;
- des taux d’échec et de rechute inférieurs (2,3 % contre 3,3 %) ; soit une réduction relative de 29 % (RR=0,71, IC 95 %: 0,12 à 3,8) ;
- des taux de décès inférieurs (0 % contre 11 %) ; soit une réduction absolue de 11 % (DR= –0.11, IC 95 % : –0.13 à –0,03) ;
- des taux de perdus de vue inférieurs (0 % contre 12 %) ; soit une réduction absolue de 12 % (DR= –0,12, IC 95 % : –0,14 à –0,04) ;
- des taux d’événements indésirables de grade 3 à 5 supérieurs (14 % contre 5 %) ; soit une augmentation relative de quatre (RRa=3,99, IC 95 % : 1,67 à 9,57) ; et
- des taux de résistance amplifiée inférieurs (0 % contre 2,4 %) ; soit une augmentation absolue de 2 % (DR=0,02, IC 95 % : –0,04 à 0,06) ;
Le GDG a estimé que les effets bénéfiques du BPaL avec le linézolide 600-26 étaient importants et que les effets indésirables étaient modérés par rapport aux schémas plus longs recommandés par l’OMS. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du BPaL avec le linézolide 600–26.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide et de linézolide (BPaL), plutôt que des schémas plus longs (18 mois), est préconisée chez les patients atteints de tuberculose MR-RR sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins d’un mois.
Sous-question PICO 5.3
Le bras BPaL 600–26 de l’essai ZeNix (dans lequel le linézolide 600 mg par jour a été utilisé pendant 26 semaines et la population comprenait des patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones) a été comparé à une cohorte de patients atteints de tuberculose MR-RR sans résistance aux fluoroquinolones traités en Afrique du Sud avec un schéma de 9 mois comprenant du linézolide pendant 2 mois. L’analyse primaire a été effectuée 12 mois après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le BPaL avec le linézolide 600-9 (n = 43) comparés aux participants atteints de tuberculose MR-RR (sans résistance aux fluoroquinolones) recevant un schéma de 9 mois avec du linézolide (n = 4 216) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (100 % contre 66 %) ; soit une augmentation relative de 52 % (RR=1,52, IC 95 % :1,38 à 1,55) ;
- des taux d’échec et de rechute inférieurs (0 % contre 1,2 %) ; soit une réduction absolue de 1 % (DR= –0,01, IC 95 % : –0,02 à 0,07) ;
- des taux de décès inférieurs (0 % contre 18 %) ; soit une réduction absolue de 18 % (DR= –0,18, IC 95 % : –0,19 à –0,1) ;
- des taux de perdus de vue inférieurs (0 % contre 15 %) ; soit une réduction absolue de 15 % (DR= –0,15, IC 95 % : –0,16 à –0,07) ;
- des taux d’événements indésirables de grade 3 à 5 supérieurs (14 % contre 4,9 %) ; soit une augmentation par trois (RRa=2.92, IC 95 % : 1,38 à 6,18) ; et
- des taux de résistance amplifiée inférieurs (0 % contre 0,6 %) ; soit une réduction absolue de 1 % (DR= –0,01, IC 95 % : –0,01 à –0,08).
Le GDG a estimé que les effets bénéfiques du BPaL avec le linézolide 600-26 étaient importants et que les effets indésirables étaient modérés par rapport au schéma de 9 mois avec le linézolide. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du BPaL avec le linézolide 600–26.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide et de linézolide (BPaL), plutôt que du schéma de 9 mois (avec le linézolide), est préconisée chez les patients atteints de tuberculose MR-RR sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins de 1 mois.
Question PICO 5 – Conclusion récapitulative intermédiaire
Les trois évaluations effectuées sous la question PICO 5 ont conduit à la formulation de la recommandation conditionnelle en faveur du schéma BPaL (600 mg – 26 semaines) par rapport au schéma de 9 mois avec l’éthionamide actuellement recommandé (sous-question PICO 5.1), aux schémas plus longs (18 mois) (sous-question PICO 5.2) et au nouveau schéma de 9 mois où l’éthionamide est remplacé par le linézolide pendant 2 mois (sous-question PICO 5.3) chez les patients atteints de tuberculose pulmonaire MR-RR sans résistance aux fluoroquinolones.
Sous-question PICO 6.1
Le bras du schéma BPaLM de l’essai TB-PRACTECAL avec une population composée de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones (tuberculose MR-RR ou pré-UR) a été comparé au bras de comparaison de l’essai TB-PRACTECAL, qui comprenait des patients atteints de tuberculose MR-RR ou pré-UR traités par plusieurs schémas de référence locaux recommandés par l’OMS au moment où l’essai a été mené (dont un schéma de 9 à 12 mois contenant des produits injectables, un schéma de 18 à 24 mois recommandé par l’OMS [avant 2019], un schéma entièrement oral de 9 à 12-mois et un schéma entièrement oral de 18 à 20 mois). L’analyse primaire a été effectuée 72 semaines après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaLM (n=62) comparés aux participants recevant les schémas de référence recommandés par l’OMS utilisés dans l’essai TB-PRACTECAL (n=66) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (89 % contre 52 %) ; soit une augmentation relative de 73 % (RRa=1,73, IC 95 % :1,31 à 2,27) ;
- des taux d’échec et de rechute inférieurs (8 % contre 26 %), soit une réduction relative de 74 % (RRa = 0,26, IC 95 % 0,10 à 0,71) ;
- des taux de décès inférieurs (0 % contre 3,0 %) ; soit une réduction absolue de 3 % (DR= –0,03, IC 95 % : –0,10 à 0,03) ;
- des taux de perdus de vue inférieurs (3,2 % contre 20 %) ; soit une réduction relative de 84 % (RR=0,16, IC 95 %: 0,04 à 0,61) ;
- des taux d’événements indésirables de grade 3 à 5 inférieurs (21 % contre 51 %) ; soit une réduction relative de 59 % (RRa =0,41, IC 95 %: 0,26 à 0,63) ; et
- des taux de résistance amplifiée inférieurs (0 % contre 1,9 %) ; soit une réduction absolue de 2 % (DR= –0,02, IC 95 % : –0,07 à –0,02).
Le GDG a estimé que les effets bénéfiques du BPaLM étaient importants et que les effets indésirables étaient négligeables par rapport aux schémas de référence recommandés par l’OMS. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du schéma BPaLM.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide, de linézolide et de moxifloxacine (BPaLM), plutôt que du schéma de 9 mois ou plus (18 mois), est préconisée chez les patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins d’un mois.
Sous-question PICO 6.2
Le bras du schéma BPaLM de l’essai TB-PRACTECAL avec une population composée de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones (tuberculose MR-RR ou pré-UR) a été comparé au bras BPaL de l’essai TB-PRACTECAL, qui comprenait des patients atteints de tuberculose MR-RR ou pré-UR. L’analyse primaire a été effectuée 72 semaines après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaLM (n=62) comparés aux participants recevant le schéma BPaL dans l’essai TB-PRACTECAL (n=60) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (89 % contre 77 %) ; soit une augmentation relative de 15 % (RRa=1,15, IC 95 % : 0,95 à 1,38) ;
- des taux d’échec et de rechute inférieurs (8,1 % contre 13 %) ; soit une réduction relative de 47 % (RRa=0,53, IC 95 %: 0,17 à 1,63) ;
- des taux de perdus de vue inférieurs (3,2 % contre 10 %) ; soit une réduction relative de 68 % (RRa =0,32, IC 95 %: 0,08 à 1,34) ;
- aucune différence dans les décès (0 % contre 0 %) ; soit une différence absolue de 0 % (DR=0, IC 95 % : –0,06 à 0,06) ;
- des taux d’événements indésirables de grade 3 à 5 supérieurs (21 % contre 20 %) ; soit une augmentation relative de 7 % (RRa=1,07, IC 95 % : 0,62 à 1,88) ; et
- des taux de résistance amplifiée inférieurs (0 % contre 2,9 %) ; soit une réduction absolue de 3 % (DR= –0,03, IC 95 % : –0,08 à –0,01).
Le GDG a estimé que les effets bénéfiques du BPaLM étaient modérés et que les effets indésirables étaient faibles par rapport au BPaL. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du schéma BPaLM.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide, de linézolide et de moxifloxacine (BPaLM), plutôt que du schéma BPaL, est préconisée chez les patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins d’un mois.
Sous-question PICO 6.3
Le bras du schéma BPaLM de l’essai TB-PRACTECAL avec une population composée de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones (tuberculose MR-RR ou pré-UR) a été comparé au bras BPaLC de l’essai TB-PRACTECAL qui comprenait des patients atteints de tuberculose MR-RR ou pré-UR. L’analyse primaire a été effectuée 72 semaines après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaLM (n=62) comparés aux participants recevant le schéma BPaLC (n = 64) dans l’essai TB-PRACTECAL (n=60) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (89 % contre 81 %) ; soit une augmentation relative de 11 % (RRa=1,1115, IC 95 % : 0,94 à 1,31) ;
- des taux d’échec et de rechute inférieurs (8,1 % contre 9,4 %) ; soit une réduction relative de 30 % (RRa =0,70, IC 95 % : 0,2 à 2,29) ;
- des taux de décès inférieurs (0 % contre 1,6 %) ; soit une réduction absolue de 2 % (DR= –0,02, IC 95 % : –0,08 à 0,04) ;
- des taux de perdus de vue inférieurs (3,2 % contre 7,8 %) ; soit une réduction relative de 59 % (RR=0,41, IC 95 %: 0,09 à 1,77) ;
- des taux d’événements indésirables de grade 3 à 5 inférieurs (21 % contre 34 %) ; soit une réduction relative de 39 % (RRa =0,61, IC 95 %: 0,37 à 1,00) ; et
- des taux de résistance amplifiée inférieurs (0 % contre 1,9 %) ; soit une réduction absolue de 2 % (DR= –0,02, IC 95 % : –0,07 à –0,02).
Le GDG a estimé que les effets bénéfiques du BPaLM étaient modérés et que les effets indésirables étaient négligeables par rapport au BPaLC. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du schéma BPaLM.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide, de linézolide et de moxifloxacine (BPaLM), plutôt que du schéma BPaLC, est préconisée chez les patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins d’un mois.
Sous-question PICO 6.4
Le bras du schéma BPaLC de l’essai TB-PRACTECAL avec une population composée de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones (tuberculose MR-RR ou pré-UR) a été comparé au bras de comparaison de l’essai TB-PRACTECAL, qui comprenait des patients atteints de tuberculose MR-RR ou pré-UR traités par plusieurs schémas de référence locaux recommandés par l’OMS au moment de l’essai (dont un schéma de 9 à 12 mois contenant des produits injectables ; un schéma de 18–24-mois recommandé par l’OMS [avant 2019] ; un schéma entièrement oral de 9 à 12 mois ; et un schéma entièrement oral de 18 à 20 mois). L’analyse primaire a été effectuée 72 semaines après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le schéma BPaLC (n=64) comparés aux participants recevant les schémas de référence recommandés par l’OMS utilisés dans l’essai TB-PRACTECAL (n=66) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (81 % contre 52 %) ; soit une augmentation relative de 55 % (RRa=1,55, IC 95 % : 1,15 à 2,11) ;
- des taux d’échec et de rechute inférieurs (9,4 % contre 26 %) ; soit une réduction relative de 66 % (RRa =0,34, IC 95 %: 0,14 à 0,87) ;
- des taux de décès inférieurs (1,6 % contre 3,0 %) ; soit une réduction relative de 48 % (RR=0,52, IC 95 %: 0,07 à 3,85) ;
- des taux de perdus de vue inférieurs (7,8 % contre 20 %) ; soit une réduction relative de 57 % (RRa =0,43, IC 95 %: 0,15 à 1,23) ;
- des taux d’événements indésirables de grade 3 à 5 inférieurs (34 % contre 51 %) ; soit une réduction relative de 33 % (RRa =0,67, IC 95 %: 0,46 à 0,97) ; et
- des taux de résistance amplifiée supérieurs (1,9 % contre 1,9 %) ; soit une augmentation relative de 4 % (RR=1,04, IC 95 % : 0,19 à 5,80) ;
Le GDG a estimé que les effets bénéfiques du BPaLC étaient importants et que les effets indésirables étaient négligeables par rapport aux schémas de référence recommandés par l’OMS. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du schéma BPaLC.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide, de linézolide et de clofazimine (BPaLC), plutôt que d’un schéma de 9 mois ou plus (18 mois), est préconisée chez les patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins d’un mois (annulée par les conclusions de sous-questions PICO 6.5 et 6.6).
Sous-question PICO 6.5
Le bras du schéma BPaLC de l’essai TB-PRACTECAL avec une population composée de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones (tuberculose MR-RR ou pré-UR) a été comparé au bras BPaL de l’essai TB-PRACTECAL, qui comprenait des patients atteints de tuberculose MR-RR ou pré-UR. L’analyse primaire a été effectuée 72 semaines après le début du traitement.
Les participants atteints de tuberculose pulmonaire MR-RR ou de tuberculose pré-UR recevant le schéma BPaLC (n = 64) comparés aux participants recevant le schéma BPaL 600-300 (n = 60) présentaient les caractéristiques suivantes :
- des taux de succès thérapeutique supérieurs (81 % contre 77 %) ; soit une augmentation relative de 4 % (RRa=1,04, IC 95 % : 0,84 à 1,30) ;
- des taux d’échec et de rechute inférieurs (9,4 % contre 13 %) ; soit une réduction relative de 14 % (RRa =0,86, IC 95 %: 0,28 à 2,69) ;
- des taux de décès supérieurs (1,6 % contre 0 %) ; soit une augmentation absolue de 2 % (DR=0,02, IC 95 % : –0,05 à 0,08) ;
- des taux de perdus de vue inférieurs (7,8 % contre 10 %) ; soit une réduction relative de 28 % (RRa =0,72, IC 95 %: 0,21 à 2,47) ;
- des taux d’événements indésirables plus élevés (34 % contre 20 %) ; soit une augmentation relative de 64 % (RRa = 1,64, IC 95 % : 0.97 à 2.79) ; et
- des taux d’amplification de la pharmacorésistance supérieurs (1,9 % contre 2,9 %) ; soit une réduction relative de 35 % (RR=0,65, IC 95 %: 0,13à 3,21).
Le GDG a estimé que les effets souhaités et les effets indésirables du BPaLC étaient faibles par rapport au BPaL. La certitude quant aux données a été jugée très faible. L’équilibre entre les effets sur la santé n’a pas permis de se prononcer en faveur du schéma d’intervention ou du schéma de comparaison ; cependant, compte tenu du coût plus élevé du schéma, d’une plus lourde charge médicamenteuse, d’une acceptabilité réduite due à la décoloration de la peau et d’autres effets indésirables potentiels liés à la clofazimine et en l’absence de nets avantages remarquables en termes d’effets sur la santé, le groupe s’est prononcé contre le schéma d’intervention.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide et linézolide (BPaL), plutôt que du schéma BPaLC, est préconisée chez les patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins d’un mois.
Sous-question PICO 6.6
Le bras BPaL de l’essai TB-PRACTECAL avec une population composée de patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones (tuberculose MR-RR ou pré-UR) a été comparé au bras de comparaison de l’essai TB-PRACTECA, qui comprenait des patients atteints de tuberculose MR-RR ou pré-UR traités par plusieurs schémas de référence locaux (dont un schéma de 9 à 12 mois contenant des produits injectables, un schéma de 18 à 24 mois recommandé par l’OMS [avant 2019], un schéma de 9 à 12 mois entièrement oral et un schéma de 18 à 20 mois entièrement oral). L’analyse primaire a été effectuée 72 semaines après le début du traitement.
Les participants atteints de tuberculose MR-RR (avec ou sans résistance aux fluoroquinolones) recevant le BPaL (n = 60) comparés aux participants recevant les schémas de référence recommandés par l’OMS utilisés dans l’essai TB-PRACTECAL présentaient les caractéristique suivantes :
- des taux de succès thérapeutique supérieurs (77 % contre 52 %) ; soit une augmentation relative de 47 % (RRa=1,47, IC 95 % : 1,09 à 1,99) ;
- des taux d’échec et de rechute inférieurs (13 % contre 26 %) ; soit une réduction relative de 48 % (RRa =0,52, IC 95 %: 0,22 à 1,18) ;
- des taux de décès inférieurs (0 % contre 3,0 %) ; soit une réduction absolue de 3 % (DR= –0,03, IC 95 % : –0,10 à 0,03) ;
- des taux de perdus de vue inférieurs (10 % contre 20 %) ; soit une réduction relative de 40 % (RRa =0,60, IC 95 %: 0,24 à 1,56) ;
- des taux d’événements indésirables inférieurs (20 % contre 51 %) ; soit une réduction relative de 62 % (RR=0,38, IC 95 %: 0,24 à 0,60) ; et
- des taux d’amplification de la pharmacorésistance supérieurs (2,9 % contre 1,9 %) ; soit une augmentation relative de 59 % (RR=1,59, IC 95 % : 0,32 à 7,84) ;
Le GDG a estimé que les effets bénéfiques du BPaL étaient importants et que les effets indésirables étaient négligeables par rapport aux schémas de référence recommandés par l’OMS. La certitude quant aux données a été jugée très faible. Le groupe a donc estimé que l’équilibre entre les effets sur la santé était probablement en faveur du schéma BPaL.
Conclusion
L’utilisation du schéma thérapeutique de 6 mois composé de bédaquiline, de prétomanide et de linézolide (BPaL), plutôt que d’un schéma de 9 mois ou plus (18 mois), est préconisée chez les patients atteints de tuberculose MR-RR avec ou sans résistance aux fluoroquinolones, qui n’ont jamais été exposés à la bédaquiline et au linézolide ou qui y ont été exposés moins d’un mois.
PICO 6 – Conclusion récapitulative intermédiaire
La principale évaluation qui a déterminé la décision globale était celle de la sous-question PICO 6.1, qui a abouti à la recommandation conditionnelle pour l’utilisation du schéma BPaLM plutôt que la combinaison interne de schémas de référence conformes aux recommandations de l’OMS sur les schémas de 9 mois ou plus longs. Les évaluations des schémas thérapeutiques expérimentaux entre eux et avec les soins de référence dans les sous-questions PICO 6.2 à 6.6 ont permis au groupe de prendre les décisions finales.
Résumé des autres données probantes
Les autres données relatives à ces questions PICO examinées par le GDG étaient une analyse coûtefficacité, une étude sur l’acceptabilité et la probabilité de la mise en oeuvre du schéma BPaL, des données pharmacocinétiques modélisées basées sur l’élaboration d’un modèle pharmacocinétiquetoxicodynamique et un résumé des données sur la toxicité pour la reproduction potentielle du prétomanide. Aucune donnée de recherche supplémentaire n’était disponible lors de l’examen des sous-questions PICO 3.2 à 3.5.
Données pharmacocinétiques
Les premières données de l’étude pharmacocinétique incluse dans l’essai TB-PRACTECAL ont été présentées au GDG lors d’un des webinaires préparatoires. Les résultats finaux de cette sous-étude n’étaient pas disponibles au moment de l’évaluation et n’ont pas pu être examinés en détail.
La pharmacocinétique du linézolide est très variable, l’efficacité et la toxicité dépendant de facteurs tels que la sensibilité aux agents pathogènes, l’exposition au médicament et l’association d’autres médicaments. La toxicité du linézolide, en particulier lorsqu’il est utilisé à des doses plus élevées et à des durées plus longues, est un phénomène connu et diverses stratégies sont préconisées pour la réduire. Cependant, à l’exception des données disponibles dans les essais ZeNix et TB-PRACTECAL, aucune autre stratégie n’a été testée dans le cadre d’un essai.¹⁷
Données sur la toxicité pour la reproduction du prétomanide
De nouvelles données relatives à l’innocuité du prétomanide fondées sur les évaluations des hormones dans quatre essais cliniques et une enquête sur la paternité ont été évaluées ; ces données ont permis d’atténuer en grande partie les précédentes inquiétudes quant aux toxicités pour la reproduction observées dans les études animales,¹⁸ indiquant que les effets indésirables sur la fécondité masculine humaine étaient peu probables. Une étude portant sur le sperme des hommes sous traitement comprenant du prétomanide est en cours et répondra à toutes les préoccupations persistantes. Ci-dessous se trouve un résumé des données précliniques et cliniques concernant la toxicité testiculaire du prétomanide :
- études toxicologiques sur les rongeurs – signes de toxicité testiculaire directe ;
- études toxicologiques sur les singes – pas de signe de toxicité testiculaire directe ; résultats de sperme anormal considérés comme une conséquence du déclin de la condition physique ;
- données sur les hormones provenant d’études cliniques – aucune modification de l’hormone folliculostimulante (FSH), l’hormone lutéinisante (LH) et l’inhibine B, correspondant à la toxicité testiculaire ;
- enquête sur la paternité – 44 enfants nés de 38 hommes (12 %) ayant participé à des études sur le prétomanide d’une durée de traitement de 4 à 6 mois ; et
- étude sur le sperme – étude en cours évaluant le sperme chez les hommes sous traitement au prétomanide.
Ressources nécessaires et rapport coût-efficacité
Les coûts estimés des schémas (chez les adultes) aux prix du Dispositif mondial pour l’approvisionnement en médicaments antituberculeux (GDF)¹⁹ sont d’environ 688 USD pour le BPaL (600–26), 716 USD pour le BPaLM (600–26), une moyenne de 771 USD pour les schémas plus longs (en fonction de la durée et de la composition) et de 535 à 557 USD pour les traitements de 9 mois. Les données de trois études étaient disponibles sur des analyses plus détaillées des ressources nécessaires et du rapport coût-efficacité ; deux de ces études comparaient le schéma BPaL à des schémas plus longs (18 mois) (26, 27) et une comparait les schémas BPaL, BPaLM et BPaLC à des schémas plus longs (18 mois) et au schéma sur 9 mois avec l’éthionamide (28). L’applicabilité des résultats de ces études variait selon les questions PICO et sous-PICO, et le groupe a émis des réserves lorsqu’il a discuté de ces résultats (les détails sont disponibles dans les tableaux décisionnels GRADE dans Annex 4). Globalement, sur la base de ces trois publications, les estimations du coût total comparatif (médicaments et livraison) dans le pays semblent être entre 1,4 et 6 fois plus élevées (schémas plus longs) ou entre 1 et 18 % plus élevées (schémas de 9 mois) que pour le BPaLM/ BPaL. Par conséquent, le groupe a estimé que la mise en oeuvre du schéma BPaLM/BPaL permettrait probablement de faire des économies importantes en remplaçant les schémas plus longs (18 mois) et des économies moyennes en remplaçant les schémas de 9 mois.
L’étude sur le rapport coût-efficacité (28) a révélé que, dans la plupart des milieux, le BPaLM/BPaL est économique, principalement en raison de la durée de soins réduite et donc de la réduction du nombre de consultations externes, de journées d’alitement et d’analyses de laboratoire. Le groupe a estimé que le rapport coût-efficacité était probablement en faveur du BPaLM/BPaL.
Équité, acceptabilité et faisabilité
Le groupe a considéré que la durée du traitement et la capacité à décentraliser le traitement (pour permettre l’accès à des milieux éloignés et mal desservis et à des populations défavorisées) avaient une incidence sur l’équité. Bien qu’il n’ait pas trouvé de données de recherche pertinentes, le groupe a fait appel à son expérience collective pour estimer que l’utilisation du BPaLM/BPaL présenterait probablement des avantages en raison de sa complexité et de sa durée réduites. Par conséquent, le groupe a estimé que l’utilisation du schéma BPaLM/BPaL renforcerait probablement l’équité.
Une étude sur l’acceptabilité et la faisabilité du schéma BPaL du point de vue des prestataires (29) a été considérée comme une donnée pertinente pour l’évaluation du BPaL et indirectement pour l’évaluation du BPaLM. Il s’agissait d’une étude sur des méthodes mixtes menée entre mai 2018 et mai 2019 en Indonésie, au Kirghizistan et au Nigeria auprès d’un échantillon représentatif d’agents de santé et de parties prenantes de programmes et de laboratoires. Les résultats de cette étude indiquaient un niveau élevé d’acceptabilité et de faisabilité. Le BPaL a été jugé acceptable par plus de 80 % des participants dans tous les domaines et parmi les parties prenantes et 88 % des parties prenantes interrogées ont déclaré qu’elles le mettraient probablement en œuvre lorsqu’il serait disponible. Les parties prenantes ont apprécié que le BPaL réduise la charge de travail ainsi que la charge financière pesant sur le système de santé ; ont exprimé des inquiétudes concernant l’innocuité du BPaL (surveillance), l’efficacité à long terme et les exigences réglementaires au niveau national ; et ont souligné l’importance de s’attaquer aux contraintes actuelles des systèmes de santé, en particulier dans les systèmes de surveillance du traitement et de la sécurité. Les résultats d’une deuxième étude qualitative (30) axée sur le point de vue des patients ont été présentés au groupe ; cette étude indiquait que les patients accueilleraient favorablement l’impact positif d’un traitement plus court sur la situation professionnelle²⁰.
Le groupe a pris note de ces résultats de l’étude et, lors de ses délibérations, a considéré les patients et les prestataires de santé comme des parties prenantes clés. Le groupe a estimé que les aspects suivants étaient essentiels en ce qui concerne l’acceptabilité du BPaLM/BPaL : durée du schéma et besoins en matière de pharmacovigilance (liés à la fois aux déplacements nécessaires, à la perte de revenus et à la perturbation générale de la vie des patients, et à la charge de travail pour le système de santé) et nécessité de DST. Le groupe a estimé que le schéma thérapeutique BPaLM/BPaL serait probablement acceptable. En ce qui concerne la faisabilité, le groupe a noté la disponibilité limitée de substances pures de médicaments dans le schéma BPaLM/BPaL à utiliser dans le DST comme obstacle potentiel à la mise en oeuvre ; il a également noté que les données sur la concentration critique de prétomanide à utiliser dans le DST étaient limitées. Toutefois, compte tenu de la durée réduite, de la complexité et de la charge de travail associée du BPaLM/ BPaL, le groupe a estimé que la mise en oeuvre du BPaLM/BPaL est probablement faisable.
14 Accessibles sur https://clinicaltrials.gov/ct2/home.
15 Protocole d’essai disponible sur https://trialsjournal.biomedcentral.com/articles/10.1186/s13063-022-06331-8.
16 Plusieurs participants ont été exclus dans chaque ensemble de données en raison d’une résistance à la rifampicine non confirmée.
17 Tel que présenté dans l’examen par des experts (par le Dr J-W. Alffenaar, Université de Sydney) au GDG dans l’un des webinaires préparatoires.
18 Il a été démontré que le prétomanide provoque une atrophie testiculaire et une altération de la fertilité chez les rats mâles
19 Les prix estimés des schémas ont été calculés à partir du prix moyen pondéré pour chaque médicament (le prix moyen pondéré tient compte des différents prix pour chaque fournisseur de ce médicament pondérés par l’allocation de parts de marché reçue de chaque appel d’offres du GDF), de la durée indiquée (en mois) et en supposant 30 jours de traitement par mois. Les coûts réels finaux peuvent varier en fonction des produits livrés.
20 Non publié, avec l’aimable autorisation de Beverley Stringer, Manson unit, Médecins sans frontières.