 Reacción
ReacciónTuberculosis Child Multidrug-Resistant Preventive Therapy Trial (TB CHAMP): Efficacy and safety of levofloxacin preventive treatment in child and adolescent HHCs of multidrug-resistant tuberculosis (MDR-TB). Author: Anneke Hesseling⁷
V-QUIN MDR-TB prevention study: Levofloxacin versus placebo for the treatment of tuberculosis infection among contacts of patients with MDR-TB. Author: Greg Fox⁸
Methods
TB CHAMP: A phase III cluster double-blind group randomized placebo-controlled trial to assess the efficacy and safety of a 6-month regimen of daily levofloxacin (6Lfx) as TB preventive treatment (TPT) in child contacts of patients with MDR-TB. The trial protocol was registered at ISRCTN registry (ISRCTN92634082; https://doi.org/10.1186/ISRCTN92634082)
V-QUIN: Double-blind parallel group randomized controlled trial to compare a 6-month regimen of daily levofloxacin (6Lfx) with placebo for the treatment of TBI. The objective was to determine the efficacy of levofloxacin (Lfx) in preventing the development of bacteriologically confirmed TB. The trial was registered prospectively with the Australian and New Zealand Clinical Trials Registry (ACTRN 12616000215426; https://anzctr.org.au/Trial/Registration/TrialReview.aspx?id=369817)
Objectives
TB CHAMP
Primary objective: To assess whether Lfx given daily for 24 weeks (15–20 mg/kg) is effective in preventing TB in child HHCs (HHC) of adults with MDR-TB
Secondary objectives
- Does Lfx have acceptable toxicity and tolerability in children?
- Is adherence similar in the study arms?
- Is Lfx cost-effective and acceptable to prevent MDR-TB in child HHC?
- Are there differences in Lfx resistance between study arms for children who develop incident TB?
V-QUIN
Primary objective: To evaluate the effectiveness of Lfx given for 6 months rather than placebo in prevention of TB disease among HHC of patients with MDR/RR-TB who have TB infection.
Secondary objectives: evaluation of:
- incidence of grade 3–4 adverse events
- mortality
- adherence to treatment (completion of > 80% of doses in < 270 days)
- cost-effectiveness
- acquired resistance to Lfx
Intervention
TB CHAMP: comparison of 24 weeks of daily Lfx (15–20mg/kg, maximum 750 mg) to 24 weeks of daily placebo.
V-QUIN: 180 days of self-administered oral Lfx, or an indistinguishable placebo, once per day. Tablets were distributed every 4 weeks, and a pill count performed at each visit. The daily dosing range was 10–15 mg/kg for adults, and 15–20 mg/kg for children, with a maximum dose of 750 mg.
Population eligibility
TB CHAMP
The study was completed in urban and rural settings of five provinces in South Africa, a high-incidence country for TB, TB/HIV and MDR/RR-TB. Children were considered eligible for enrolment if they fulfilled all the inclusion criteria and none of the exclusion criteria, as defined below.
Criteria for inclusion of a child or adolescent participant
- child or adolescent < 18 years who is a HHC of an adult MDR-TB index case (as stated under adult MDR-TB eligibility criteria) (up to version 2.0 protocol, only children aged < 5 years were eligible)
- primary residence in the household of the adult MDR-TB index patient or any contact resulting in significant exposure of the child
- consent from the parent or legal guardian for HIV testing
- consent from the parent or legal guardian for enrolment
- assent obtained from any child or adolescent ≥ 7 years
- if > 5 years and < 18 years of age, the child or adolescent must have a positive IGRA (Quantiferon-Gold Plus, Qiagen) test before enrolment, unless HIV positive. Children < 5 years eligible regardless of IGRA status. All HIV-positive children < 18 years of age are eligible regardless of IGRA test status.
Criteria for exclusion of a child or adolescent participant
- TB disease at enrolment
- currently on INH or a fluoroquinoline (e.g. Lfx, moxifloxacin, ofloxacin or ciprofloxacin) for ≥ 14 days. TPT may be interrupted provided that the child or adolescent participant is recruited into the study as soon as possible.
- treated for TB in the previous 12 months
- known concurrent exposure to an INH-susceptible (including rifampicin [RIF] mono-resistant) index case
- weight < 3.0 kg
- positive pregnancy test at enrolment (For women who become pregnant on study, continuation on study treatment is allowed.)
- ≤ 6 months post-partum
Inclusion criteria for adult index patients
- age ≥ 18 years
- bacteriologically confirmed pulmonary TB diagnosed from a sputum sample, treatment for MDR-TB started within the preceding 6 months
- genotypic or phenotypic resistance to INH and rifampin (RIF). If only tested by Xpert MTB/RIF or MTB/RIF Ultra or other approved molecular tests e.g. line probe assay, the index case can be included if RIF-resistant, without other confirmed DST; i.e. confirmation of both RIF and INH resistance not required.
- written informed consent from the index case (or a close relative if the index case is deceased prior to the completion of screening)
- at least 1 HHC below the age of 18 years reported to have been residing in the same household as the adult index case in the previous 6 months
Exclusion criteria for adult index patients
- MDR-TB with confirmation of genotypic or phenotypic resistance to fluoroquinolones (FQs) (version 3.0 protocol)
V-QUIN
The study was conducted in Viet Nam, which is among the high-incidence countries for TB and MDR/RR-TB. Participants were recruited in urban and rural settings in 10 provinces. The study sites delivered standard programmatic management of drug-resistant TB within the National Tuberculosis Programme (NTP).
Inclusion criteria for randomization
- all ages (participants < 15 years were enrolled only during the final 6 months of recruitment in conformity with the requirements of the local institutional review board)
- either:
- tuberculin skin test (TST) positive, defined as either (a) ≥ 10 mm first reading; (b) new TST conversion on the second reading (≥ 10 mm at second reading and an increase of ≥ 6 mm at the second reading over the first reading, OR
- any TST size if known HIV positive or severely malnourished (body mass index < 16 kg/m2 ).
Exclusion criteria
- current TB disease
- known to be pregnant
- unable to take oral medication
- body weight < 3 kg
- unwilling or unable to participate in follow-up
- currently breastfeeding
- known allergy to FQ antibiotics or history of severe tendinopathy related to FQs
- currently taking another medication reported to increase the cardiac QTc interval
- documented previous treatment for MDR-TB
- documented treatment with antibiotics that are active against MDR-TB in the previous months
- prior severe blistering reaction to tuberculin
- end-stage liver failure (class Child-Pugh C)
- dialysis-dependent chronic kidney disease
- a baseline liver function test, aspartate or alanine aminotransferase over three times the upper limit of normal
- kidney tests show end-stage kidney disease (estimated glomerular filtration rate < 20 mL/min)
- platelet count < 50 × 109 cells/L
- baseline electrocardiogram shows a QT segment > 450 ms (adults)
Randomization and trial procedure
TB CHAMP: All eligible children in a household were treated with the same drug (either all Lfx or all placebo). Households were randomized (allocated by chance) to be in the Lfx or the placebo group. Allocation conducted by computer, and households had an equal chance of being in either group. In this “double blind” study, neither the children (or their family) nor the researchers knew whether the tablets each child took were Lfx or placebo.
A CXR (anteroposterior and lateral images) was completed at baseline and, if any evidence of TB on the CXR or if the child had any symptoms or signs suggestive of TB, they underwent sampling for mycobacterial evaluation. IGRA and HIV testing were done in all children at baseline, and the result was required before enrolment of children aged 5–17 years. A pregnancy test was performed at baseline for all female participants who had begun menstruation. A full blood count, alanine transaminase and bilirubin were collected at baseline in all children. Children were followed at 4, 8, 12, 16, 24, 48 and 72 weeks and at additional unscheduled visits as clinically indicated. At each visit, children were assessed clinically for symptoms and signs of TB, new exposure to TB and for evidence of any adverse events due to the medication. Adherence to medication was quantified by pill returns and counts, treatment diaries and questionnaires. Weight and height were measured at each visit, all concomitant medications documented, and any outpatient or inpatient health-care visits were recorded. The dose of medication was adjusted monthly as necessary. A CXR (AP and lateral) was done at baseline and at 12 and 48 weeks and at any time of clinical concern. Two respiratory samples were collected for mycobacterial evaluation if the child had any symptoms or signs suggestive of TB or if they had an abnormal CXR. Sampling for presumed pulmonary TB consisted of induced sputum or gastric aspiration in children < 5 years, while children aged ≥ 5 years were encouraged to produce an expectorated sputum sample. Samples for presumed extrapulmonary TB were taken according to the site of disease. All samples were examined by smear microscopy, Xpert MTB/RIF Ultra and mycobacterial culture. Drug susceptibility (first- and second-line drugs) was tested in all mycobacterial isolates by genotypic and phenotypic methods.
V-QUIN: Participants were assigned to parallel groups in a 1:1 ratio in a permuted block design with varying block size, stratified by province. The allocation sequence was concealed before randomization. Within a household, participants were placed on the same regimen if enrolled within 90 days of one another to avoid a contamination effect.
During the 6-month treatment period, participants attended a clinic monthly to assess toxicity and support adherence. Patients were also telephoned between scheduled visits, every 2 weeks. After treatment, participants attended follow-up sessions for assessment of incident TB with a symptom screen and CXR at 6, 12, 18, 24 and 30 months. In addition, patients were assessed by telephone interviews for symptoms every 3 months during the follow-up. Throughout follow-up, participants with symptoms consistent with TB or CXR abnormalities were asked to produce three sputum samples for Xpert MTB/RIF and liquid culture. After the 30-month follow-up period (up to 134 weeks), participants were asked to produce a single sputum sample for Xpert MTB/RIF testing. Those diagnosed with TB disease were treated with a standard first- or second-line regimen according to national guidelines and the drug susceptibility profile – if available.
Outcome ascertainment
TB CHAMP: The primary end-point for efficacy was incident TB disease (bacteriologically confirmed or clinically diagnosed) or death from TB at the 48-week study visit after randomization, with a 6-week window allowed, i.e. through week 54. The prespecified main secondary end-point for safety was adverse events (AEs) grade ≥ 3 assessed by the site investigator as at least possibly associated with the study treatment. Other secondary end-points included:
- TB disease by 72 weeks
- all-cause mortality
- any AEs grade ≥ 3 from starting study treatment up to 30 days after the last study drug dose
- serious AEs up to 30 days after the last study drug dose
- discontinuation of study treatment due to AE(s)
- selected pre-defined AEs, from starting treatment up to 30 days after the last study drug dose unless stated otherwise (arthritis, arthralgia, tendinopathy during overall study follow-up, peripheral neuropathy, central nervous system effects, severe rash/cutaneous reaction and drug related fever)
- treatment adherence.
Incident TB and cause of death were adjudicated by an independent end-point review committee who were unaware of the randomized treatment allocation, according to all available clinical, radiological, microbiological and molecular data according to standard international consensus case definitions.
V-QUIN: Outcomes were reported for each participant. The primary study end-point was bacteriologically confirmed TB, defined as a positive identification of M. tuberculosis by culture or a molecular WHO-recommended rapid diagnostic in a close contact with clinical and/or radiological evidence of TB disease. The primary outcome was assigned by an expert clinical panel that was blinded to group allocation.
Secondary end-points included all forms of TB (bacteriologically confirmed or clinically probable), completion of therapy, treatment discontinuation due to an adverse event, grade 3 or 4 adverse events, death from any cause except violence, accident or acquired resistance to FQs in comparison with the index isolate. Completion of treatment was defined as having taken at least 80% of doses within 270 days after starting therapy. Secondary safety outcomes were assigned by an expert clinical panel that was blinded to group allocation.
Statistical methods
TB CHAMP
Sample size
In the original sample size calculations, a 50% reduction in TB disease incidence was assumed by 48 weeks (i.e. 50% efficacy of Lfx), from 7% in the control group to 3.5% in the Lfx group. The originally calculated sample size was 1556 participants, which would provide 80% power for the study at a 5% two-sided significance level, assuming an average of two participants enrolled per household; the household intra-class correlation was 0.10, with 10% loss to follow-up. In May 2019, after discussion with the Trial Steering Committee and the Independent Data Monitoring Committee, the target sample size was reduced to 1009 according to an assumed efficacy of 60% for Lfx (with other assumptions remaining unchanged). This assumption was considered to be in line with the results of the meta-analysis by Marks et al. (2017, doi:10.1093/cid/cix208).
Statistical analyses
- primary efficacy analysis included all randomized participants except for any late screening failures due to TB at baseline (mITT population);
- pre-defined ± 6-week window allowed for study visit at 48 weeks, with follow-up time censored at 54 weeks;
- Cox regression used to estimate hazard ratio of time to TB end-point with Lfx compared with placebo, accounting for household clustering and adjusting for site and age group;
- safety analyses included all randomized participants who had received at least one dose of study drug and comparison of time to first event between treatment arms; and
- IPD and Bayesian analysis: TB-CHAMP and V-QUIN
V-QUIN
Sample size
The risk of incident TB in the placebo group was expected to be 3% during the follow-up period, with an expected reduction in incident TB with Lfx by 70% in the treatment group, based on estimates of isoniazid efficacy in DS-TB. The sample size was increased to allow for 17% FQ resistance among patients with RR/MDR-TB in Viet Nam, a 10% drop-out rate and a design effect of 1.04 at district level and 1.07 at household level. To determine superiority, the required sample size was 1003 per arm on the basis of a two-sided alpha level of 0.05 and a power of 80%, allowing for clustering at district and household levels.
Statistical analyses
- The analysis was conducted according to a plan. Group assignment was blinded until analyses were complete. The primary analysis included the intention-to-treat (ITT) population. ITT analyses were also performed on the secondary (composite) outcomes of bacteriologically confirmed or clinically probable TB and all-cause mortality. The per-protocol population included all randomized participants who completed at least 80% of their assigned treatment. The mITT population excluded contacts of patients with RIF-susceptible TB and participants who did not start therapy.
- An interim safety analysis was performed to assess the rate of grade 3 and 4 adverse events after 600 contacts had completed 6 months of therapy. A pre-specified secondary Bayesian analysis was conducted to evaluate the incidence of confirmed or clinically probable TB at 54 weeks.
- The incidence rate ratios and 95% confidence intervals (95% CIs) were estimated in a marginal Poisson regression model fitted via generalized estimating equations.
- A complete case analysis was performed for the primary and secondary analyses.
Fig. A5.1. CONSORT diagram
TB CHAMP: Overview of enrolment, randomization and analysis of multi-drug-resistant tuberculosis child HHCs
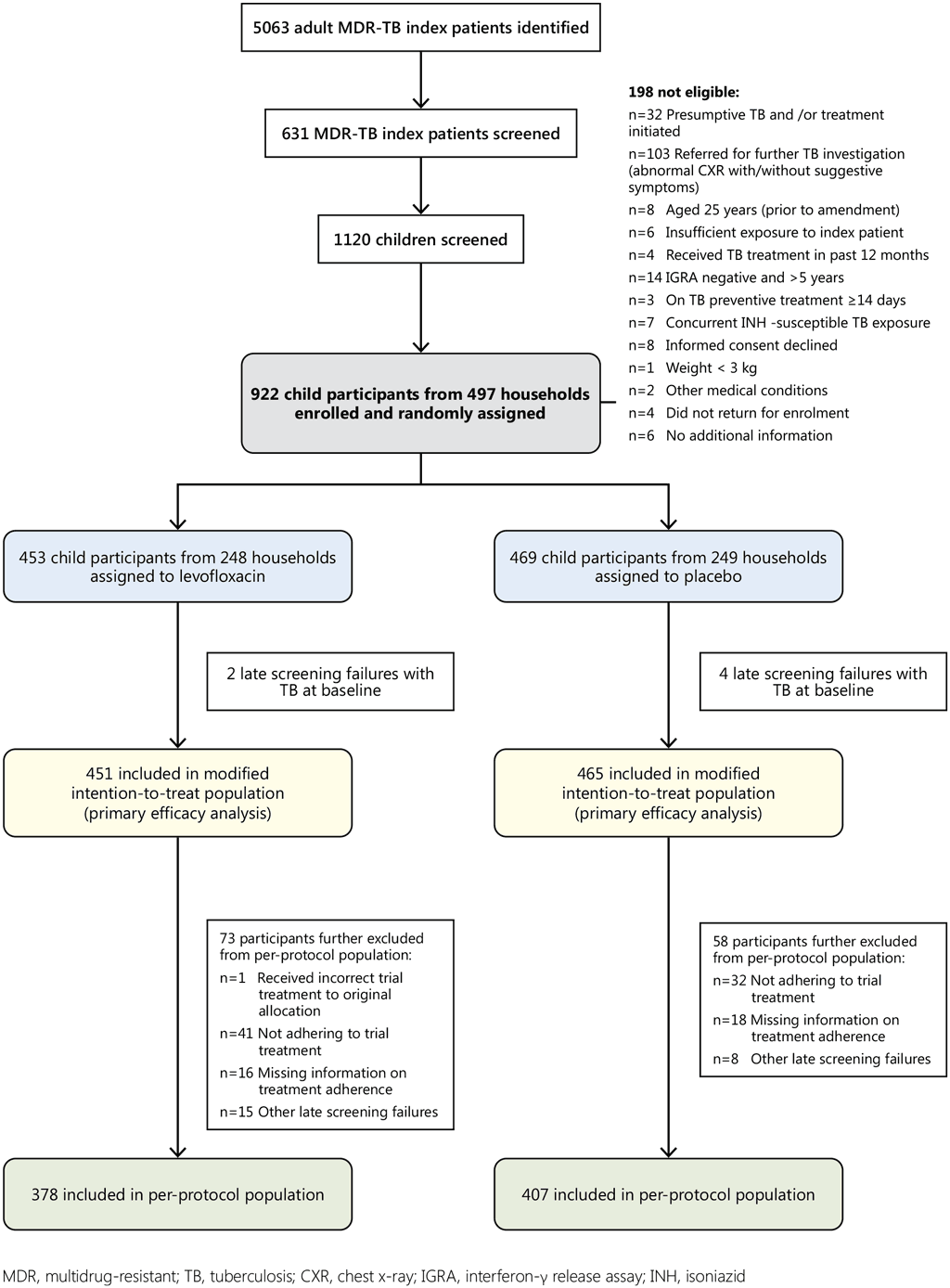
Of the 5063 adult index patients identified, 631 were screened. Index patients were ineligible for screening because: the study team was unable to establish contact with the index patient, the index patient had died or moved, the index patient was < 18 years of age, the index patient had RIF-mono-resistant TB, had been on treatment for more than 6 months, had non-pulmonary TB, or no children < 5 years were reported to be living in the household during the past 6 months.
Fig. A5.2. V-QUIN: recruitment of HHCs
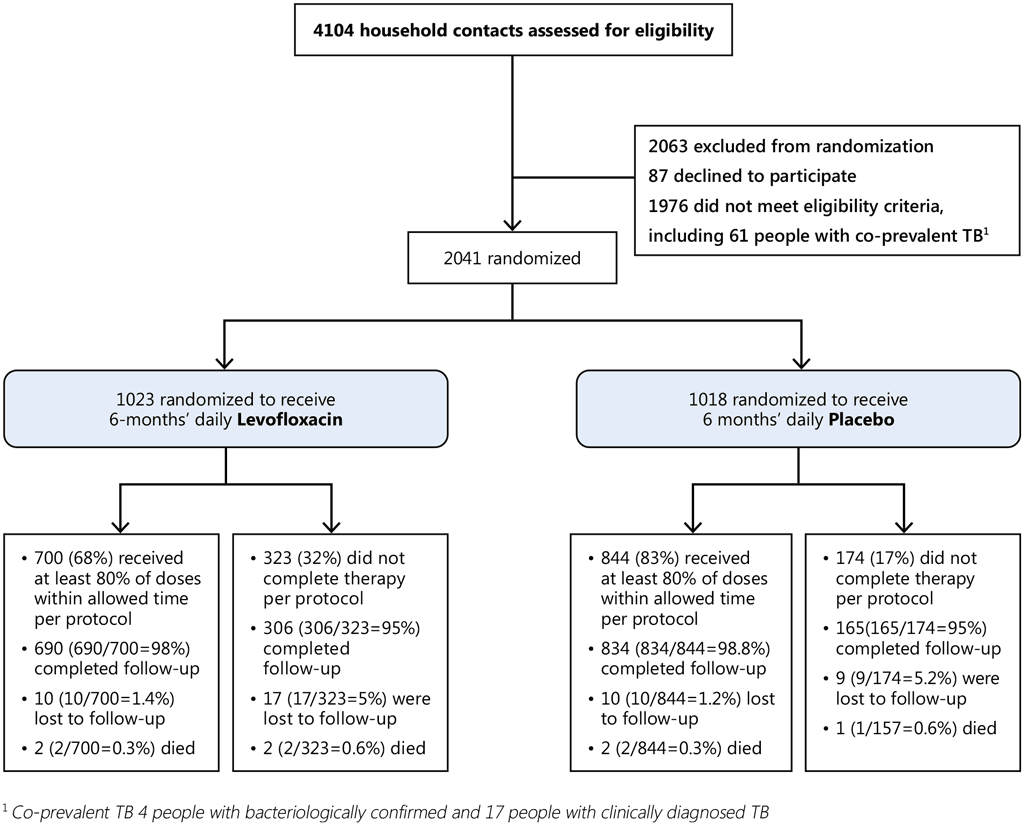
Results
TB CHAMP
Table A5.1. Baseline characteristics of randomized child participants (N=922)
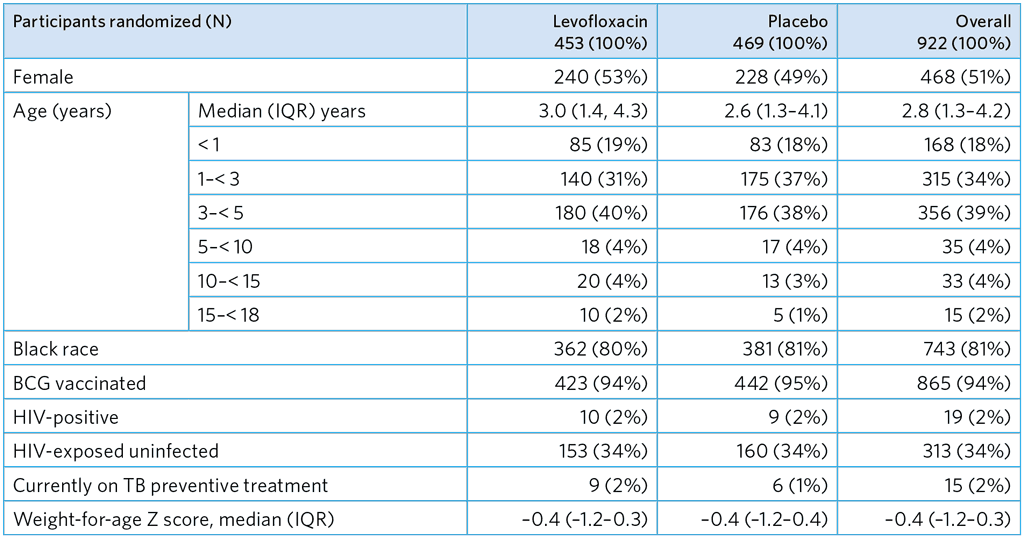
Children and adolescents aged 5–17 years were required to be IGRA-positive or living with HIV to be eligible
BCG, bacille Calmette-Guérin; IQR, interquartile range
Table A5.2. Primary efficacy analysis – mITT population
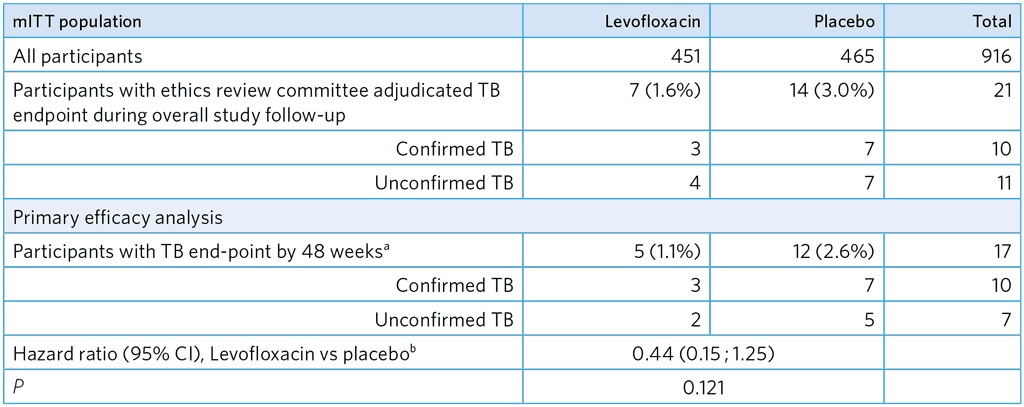
a Allowing for pre-defined ± 6–week window at study visit at 48 weeks
b Hazard ratio estimated by adjusting for site, age group and allowing for household clustering
mITT, modified intention-to-treat
Table A5.3. Primary safety analysisa
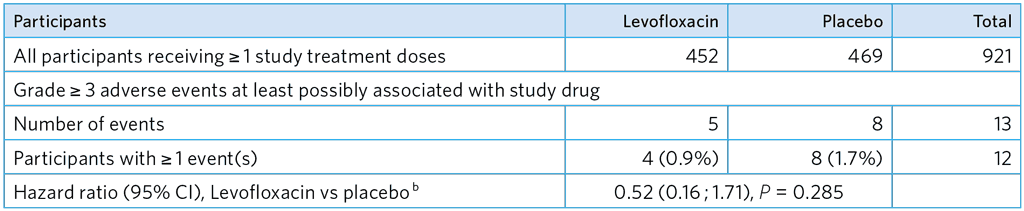
a Analyses based on time to first event.
b Hazard ratio estimated adjusting for site, age group and allowing for household clustering.
Table A5.4. All-cause mortality

a As adjudicated by the ERC.
Results V-QUIN
Table A5.5. Participant characteristics
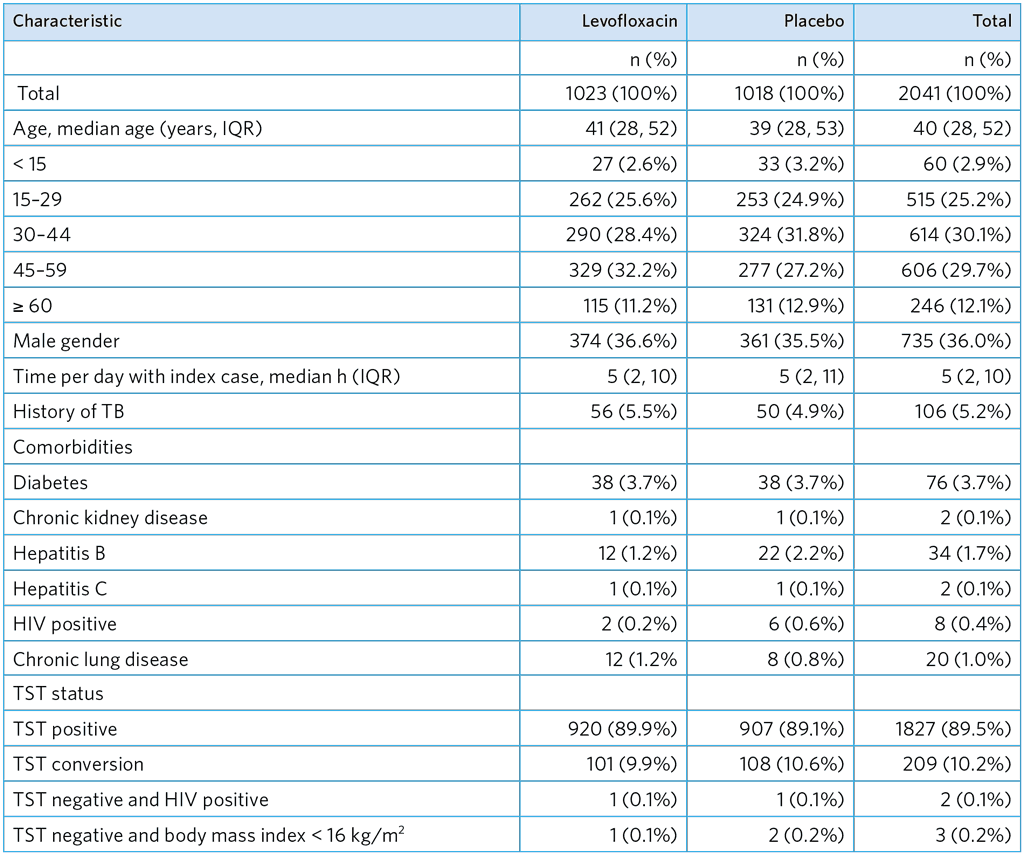
IQR, interquartile range; TST, Tuberculin skin test
Table A5.6. Incidence of TB among all participants
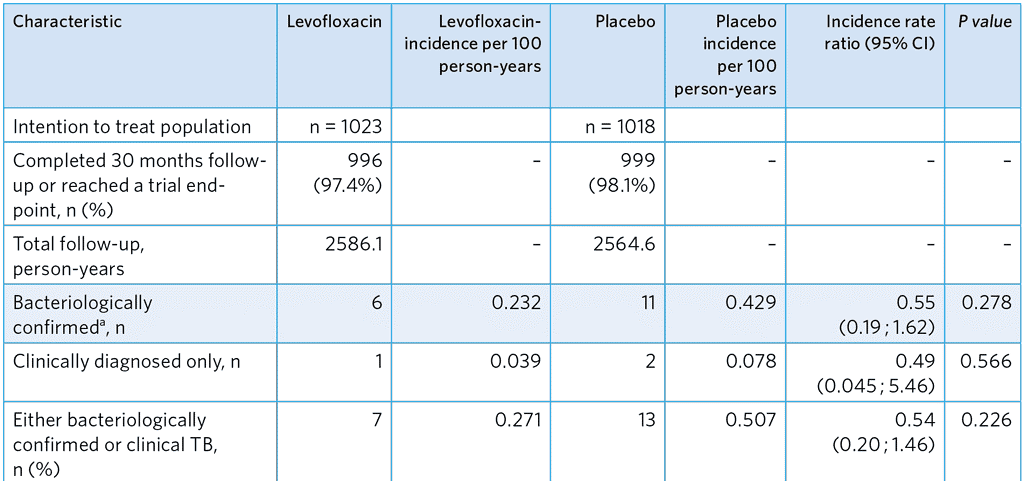
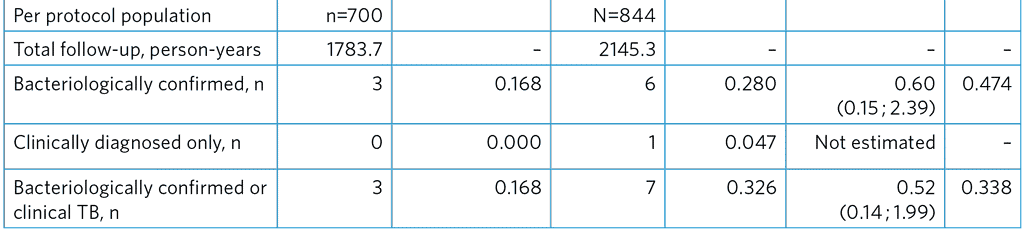
a Primary effectiveness outcome
Table A5.7. Adverse events (intention to treat population), per subject
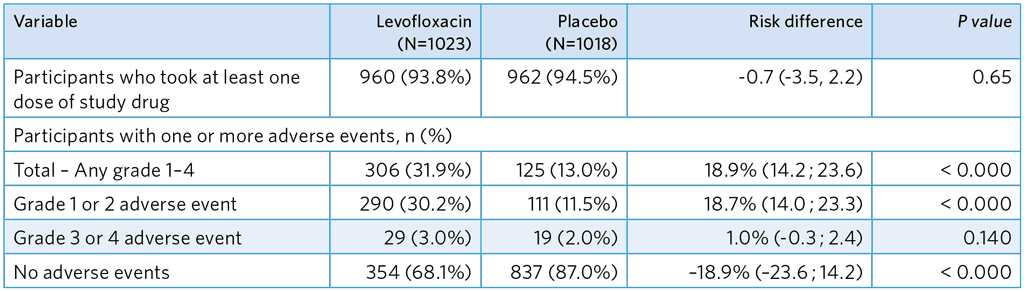
Secondary safety outcome shown in the shaded row, grade 3–4 adverse events were graded by a blinded expert clinical panel.
Table A5.8. Deaths occurring during and after the treatment period
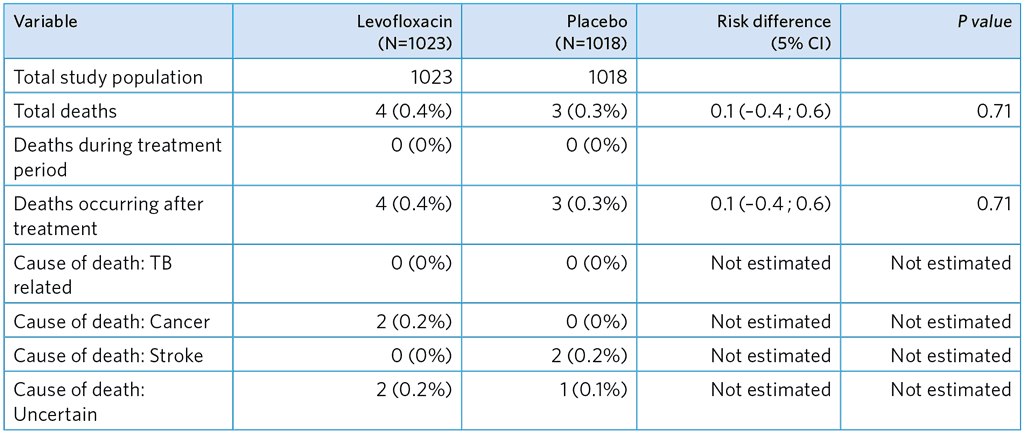
Cause of death assigned at verbal autopsy conducted at completion of the study follow-up period
Table A5.9. TPT completion
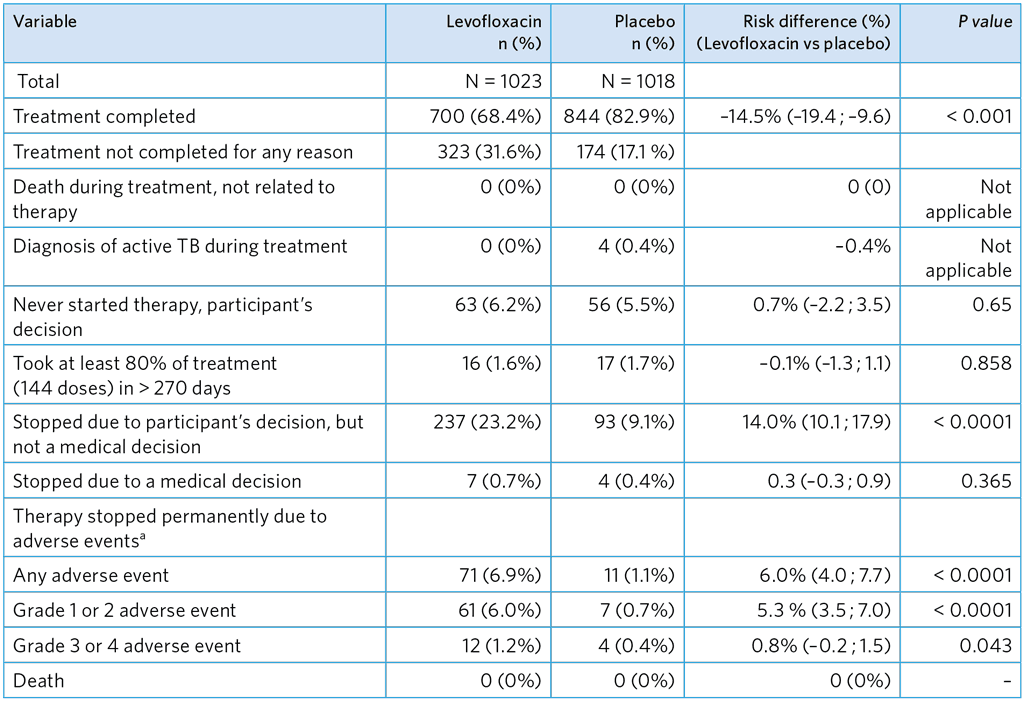
a One participant stopped due to both a grade 3–4 and grade 1–2 adverse events.
Overall trial conclusions
TB CHAMP
- Evidence of Lfx efficacy with substantial effect size: 1.1% in Lfx arm vs 2.6% in placebo arm (HR, 0.44 [95% CI 0.15 ; 1.25])
- Stronger evidence of treatment effect in site-assessed end-points and Bayesian analysis
- Lfx extremely safe in children: only 6 children in Lfx arm discontinued treatment early due to AEs compared, with 1 in the placebo arm
- Rate TB end-points lower than expected
- A high proportion of children were screened out with presumptive TB
- Lower IGRA positivity than expected. Power calculation assumed 40%+ vs 20%.
V-QUIN
- Lfx associated with a 45% reduction in microbiologically confirmed incident TB at 30 months.
- Few event resulted in broad confidence limits in the primary analysis, which spanned the null (not statistically significant)
- The incidence of grade 3–4 AEs was low, and no difference was seen between groups
- No acquired drug resistance to Lfx was observed
- About three times as many co-prevalent as incident TB cases
- In a sub-study, microbiome diversity was persistently reduced 6 months after therapy, with an increase in nasal carriage of FQ-resistant Staphylococcus aureus, a type associated with greater virulence
7 Stellenbosch University, Cape Town, South Africa
8 University of Sydney, Sydney, Australia