 Reacción
ReacciónEl grupo de elaboración de las directrices del 2018 evaluó la contribución individual a los resultados de los pacientes de los medicamentos utilizados en esquemas alargados contra la TB-MDR, utilizando principalmente las estimaciones del efecto del metanálisis del conjunto de datos de pacientes individuales del 2018 y el ensayo 213 (delamanid) para la pregunta PICO 3-2018 (TB-RR/MDR, 2018) (véanse en el anexo 3 en la web los respectivos resúmenes GRADE de la evidencia para cada fármaco, y en el anexo 4 en la web el marco para pasar de los datos a las decisiones). Tras una evaluación exhaustiva de los beneficios y daños relativos, se formularon recomendaciones para cada fármaco y se clasificaron en tres grupos (véanse los cuadros 3.1, cuadros 3.2 y cuadros 3.3).
- Grupo A: las fluoroquinolonas (levofloxacina y moxifloxacina), la bedaquilina y el linezolid se consideraron muy eficaces, y se hizo una recomendación firme para que se incluyan en todos los esquemas, salvo que estén contraindicados.
- Grupo B: la clofazimina y la cicloserina o la terizidona se recomendaron de manera condicional como fármacos de segunda línea.
- Grupo C: incluye el resto de fármacos que se pueden utilizar cuando no es posible configurar un esquema con fármacos de los grupos A o B. Los medicamentos del grupo C se clasifican según el balance relativo de beneficios y daños que suelen preverse de cada uno de ellos.
Otros medicamentos que no están incluidos en los grupos A-C son los siguientes:
- Kanamicina y capreomicina: cuando se utilizaron estos fármacos, se asociaron a peores resultados; por lo tanto, ya no se recomienda su uso en esquemas contra la TB-MDR.
- Gatifloxacina, isoniacida en dosis altas y tioacetazona: la gatifloxacina y la isoniacida en dosis altas se utilizaron tan solo en unos cuantos pacientes y la tioacetazona no se utilizó en absoluto. Actualmente no se dispone de preparados de gatifloxacina de calidad garantizada, tras su retirada del mercado debido a problemas de disglucemia. Es poco probable que la tioacetazona desempeñe un papel en los esquemas alargados contemporáneos; hoy en día no está disponible en una formulación de calidad garantizada. La isoniacida en dosis altas puede ser útil en los pacientes con TB con sensibilidad confirmada a la isoniacida (véase la sección 3.4).
- Ácido clavulánico: debe incluirse en los esquemas contra la TB-RR/MDR únicamente como fármaco complementario de los carbapenémicos (imipenem-cilastatina y meropenem). Cuando se use de esta manera, debe administrarse con cada dosis del fármaco carbapenémico, y no hay que contarlo como un medicamento eficaz adicional contra la TB.
No fue posible formular ninguna recomendación sobre la perclozona, el interferón o el sutezolid, debido a la falta de datos definitivos sobre los resultados del tratamiento obtenidos en estudios adecuados en pacientes.
En lo que respecta al uso de la bedaquilina en pacientes menores de 18 años, y considerando que los perfiles de la relación entre exposición y respuesta (eficacia) pueden extrapolarse de los pacientes adultos a los pediátricos, el grupo de elaboración de las directrices llegó a la conclusión de que las dosis evaluadas en la población infantil y adolescente en dos ensayos (ensayo de fase II TMC207-C211 y ensayo de fase I-II IMPAACT P1108; véase el anexo 5 en la web) no parecen dar lugar a exposiciones que impliquen un riesgo mayor de fracaso del tratamiento en los pacientes de 6-17 años. El riesgo de toxicidad en la población infantil de 6 años en adelante incluida en los ensayos —en todos los casos sin infección por el VIH y con escasa exposición a otros fármacos que prolongan el intervalo QT— no fue mayor que el de la población adulta. La variabilidad presente en esa muestra de tamaño reducido impidió hacer comentario alguno sobre la relación entre exposición y respuesta (seguridad). El grupo de elaboración de las directrices del 2018 también llegó a la conclusión de que las consideraciones respecto al balance de riesgos y beneficios del uso de la bedaquilina en pacientes de 6-17 años son similares a las de la población adulta, pero hizo hincapié en la necesidad de contar con más datos antes de plantearse reclasificar esta recomendación como recomendación “firme”.
En la revisión del grupo de elaboración de las directrices del 2021 se determinó que el balance entre los efectos deseables y los indeseables probablemente favorecía al uso de la bedaquilina en los menores de 6 años. El grupo de elaboración de las directrices del 2021 destacó que los beneficios pueden variar en función de contextos específicos y de características del grupo poblacional, como el estado nutricional. El grupo de elaboración de las directrices también observó que el costo potencialmente mayor de la bedaquilina en un esquema de tratamiento contra la TB-RR/MDR debería considerarse en el contexto de los beneficios de los esquemas acortados sin medicamentos inyectables (a saber, menos desplazamientos, menos tiempo de permanencia en los consultorios y menos eventos adversos). Además, el grupo consideró que la equidad podría mejorar cuando la bedaquilina esté disponible para la población infantil de corta edad, dado que su uso sería aceptable para la mayoría de las partes interesadas y que uno de los principales aspectos de la viabilidad estaría relacionado con la necesidad de farmacovigilancia (es decir, el acceso al seguimiento del ECG, así como la capacitación del personal para el seguimiento). En cualquier caso, el grupo de expertos consideró que la introducción del uso de la bedaquilina en la población infantil de corta edad era probablemente factible.
Con respecto al uso del delamanid en menores de 6 años, en la revisión del grupo de elaboración de las directrices del 2018 se decidió que, teniendo en cuenta los resultados obtenidos en la población adulta y los datos farmacológicos y de seguridad revisados, las extrapolaciones relativas a la eficacia y la seguridad deberían restringirse a la población infantil de 3-5 años, pero no a la menor de 3 años (véase el anexo 5 en la web ). Los perfiles de exposición en la población infantil de 3-5 años fueron semejantes a los de la población adulta y no fueron superiores a los de la población infantil de 6 años en adelante, para quienes los anteriores grupos de elaboración de directrices convocados por la OMS ya habían recomendado el uso del delamanid (10, 64). Sobre la base de los datos de laboratorio y cardíacos proporcionados, no se observaron en la población infantil de 3-5 años señales de toxicidad distintas a las notificadas en la población adulta. No obstante, al grupo de elaboración de las directrices le preocupaba la viabilidad de administrar la dosis correcta a la población de 3-5 años, dado que la formulación especial utilizada en el ensayo (25 mg) no estaría disponible en un futuro previsible, y que solo se dispone del comprimido para la población adulta (50 mg), que no es bioequivalente y presenta dificultades para manipular su contenido sin comprometer su eficacia.
En la revisión del grupo de elaboración de las directrices del 2021 se llegó a la conclusión de que el balance entre los efectos deseables y los indeseables probablemente favorecía al uso del delamanid en la población menor de 3 años. El grupo de elaboración de las directrices del 2021 afirmó, además, que cuando el comprimido dispersable de 25 mg estuviera disponible en el futuro, podrían variar las implicaciones en materia de recursos. Se pensó que los esquemas alargados de tratamiento que contienen delamanid podrían mejorar la equidad y ser aceptables para las partes interesadas. Además, el grupo de elaboración de las directrices del 2021 afirmó que el uso de delamanid en la población infantil de todas las edades probablemente sería factible, en especial porque se preveía que la formulación de delamanid adaptada a los pacientes pediátricos estuviera disponible a finales del 2021 (esta presentación ya está disponible). En esta declaración también se consideró que los comprimidos para la población adulta no pueden dividirse, triturarse ni disolverse para facilitar su administración a la población infantil sin que exista la posibilidad de que se altere su biodisponibilidad.
Como resultado de estas múltiples revisiones, y a medida que se ha ido disponiendo de nuevos datos, el uso de la bedaquilina y del delamanid ya no está restringido por la edad del paciente.
Cuadro 3.1 Agrupación de los fármacos recomendados para su uso en esquemas alargados contra la TB MDRᵃ
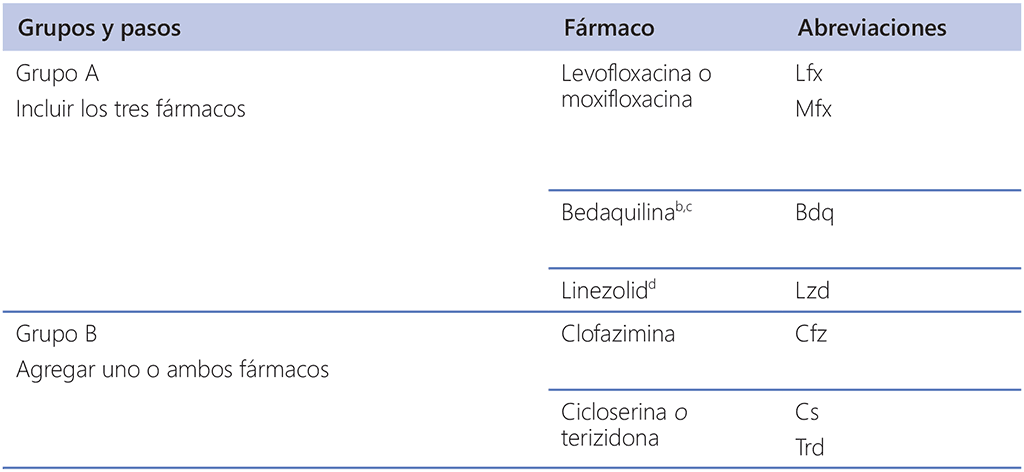


TB-MDR: tuberculosis multirresistente; TB: tuberculosis.
a El propósito de este cuadro es orientar el diseño de esquemas alargados de tratamiento individualizados contra la TB-MDR (la composición del esquema acortado de tratamiento recomendado contra la TB-MDR está ampliamente estandarizada; véase la sección 2). Los fármacos del grupo C se clasifican por orden decreciente de preferencia habitual de uso, sujeto a otras consideraciones. El metanálisis de datos de pacientes individuales del 2018 relativo a esquemas alargados no incluyó a pacientes tratados con tioacetazona e incluyó a un número demasiado bajo de pacientes tratados con gatifloxacina y con isoniacida en dosis altas para que el análisis fuera significativo. No fue posible formular ninguna recomendación sobre la perclozona, el interferón o el sutezolid debido a la falta de datos finales sobre el resultado del tratamiento obtenidos en estudios adecuados (véase el anexo 5 en la web).
b Se suele administrar la bedaquilina en dosis de 400 mg por vía oral una vez al día durante las primeras 2 semanas, seguido por 200 mg por vía oral tres veces por semana durante 22 semanas (duración total de 24 semanas). Como resultado de múltiples revisiones, y a medida que se ha ido disponiendo de nuevos datos, el uso de la bedaquilina y del delamanid ha dejado de estar restringido por la edad del paciente. La evidencia sobre la seguridad y la eficacia del uso de la bedaquilina durante más de 6 meses fue insuficiente para la revisión del 2018. Por consiguiente, el uso de la bedaquilina durante más de 6 meses se adoptó de conformidad con las prácticas óptimas para el uso de medicamentos “en indicaciones no autorizadas” (65). En el 2019, el grupo de elaboración de las directrices contó con nueva evidencia sobre el perfil de seguridad de la bedaquilina utilizada durante más de 6 meses, pero no pudo evaluar el impacto de su uso prolongado en la eficacia, debido a lo limitado de la evidencia y a la posible confusión residual de los datos. Sin embargo, la evidencia confirma que es seguro utilizar la bedaquilina durante más de 6 meses en pacientes sometidos a programas apropiados de vigilancia al inicio y durante el seguimiento. El uso de la bedaquilina durante más de 6 meses sigue siendo una indicación no autorizada y, a este respecto, siguen siendo pertinentes las prácticas óptimas para el uso en tales indicaciones.
c La evidencia sobre el uso simultáneo de bedaquilina y delamanid fue insuficiente para la revisión del 2018. En el 2019, el grupo de elaboración de las directrices contó con nueva evidencia sobre el uso simultáneo de la bedaquilina y del delamanid. Con respecto a la seguridad, el grupo de elaboración de las directrices llegó a la conclusión de que los datos no parecen indicar ningún otro problema de seguridad relativo al uso simultáneo de bedaquilina y delamanid. Ambos pueden utilizarse simultáneamente en los pacientes que tienen pocas opciones de tratamiento aparte de estos fármacos, siempre que se haga un seguimiento suficiente (que incluya un ECG y la vigilancia de los electrolitos al inicio y durante el tratamiento). El grupo de elaboración de las directrices examinó los datos sobre la eficacia del uso simultáneo de bedaquilina y delamanid; sin embargo, debido a lo limitado de la evidencia y a la posible confusión residual de los datos, no pudo formular una recomendación sobre la eficacia.
d Se demostró que el uso del linezolid durante al menos 6 meses aumenta la eficacia, aunque la toxicidad puede limitar su uso. Según indicó el análisis, el uso de linezolid durante todo el tratamiento optimizaría su efecto (alrededor del 70% de los pacientes tratados con linezolid en los que se disponía de datos lo recibieron durante más de 6 meses, y el 30% durante 18 meses o durante todo el tratamiento). No se pudieron deducir factores predictivos de la suspensión prematura del tratamiento con linezolid a partir del subanálisis de datos de pacientes individuales.
e La evidencia sobre la seguridad y la eficacia del uso del delamanid durante más de 6 meses fue insuficiente para la revisión. El uso del delamanid más allá de estos límites debería seguir las prácticas óptimas para el uso en “indicaciones no autorizadas” (65). Como resultado de múltiples revisiones, y a medida que se ha ido disponiendo de nuevos datos, el uso del delamanid ha dejado de estar restringido por la edad del paciente.
f La pirazinamida se cuenta como un fármaco eficaz únicamente cuando los resultados de las pruebas de sensibilidad a fármacos confirman la sensibilidad.
g Todas las dosis de imipenem-cilastatina y de meropenem se administran con ácido clavulánico, que solo está disponible en formulaciones en combinación con amoxicilina. La combinación de amoxicilina y ácido clavulánico no se cuenta como un medicamento eficaz adicional contra la TB ni debe usarse sin administrar imipenem-cilastatina o meropenem.
h Solo se considerará el uso de la amikacina o la estreptomicina si los resultados de las pruebas de sensibilidad a fármacos confirman la sensibilidad y si se puede garantizar el seguimiento de la pérdida de audición mediante audiometrías de gran calidad. Únicamente se planteará el uso de la estreptomicina si no se puede utilizar la amikacina (es decir, si no se dispone de ella o en caso de resistencia documentada) o si los resultados de las pruebas de sensibilidad a fármacos confirman la sensibilidad (la resistencia a la estreptomicina no se puede detectar mediante pruebas moleculares con sondas lineales de segunda línea, y es necesario realizar pruebas fenotípicas de sensibilidad a fármacos). Ya no se recomienda usar la kanamicina ni la capreomicina en los esquemas contra la TB-MDR.
i Estos fármacos fueron eficaces solo en esquemas sin bedaquilina, linezolid, clofazimina o delamanid y, por lo tanto, se proponen únicamente cuando no haya otras opciones para configurar un esquema.
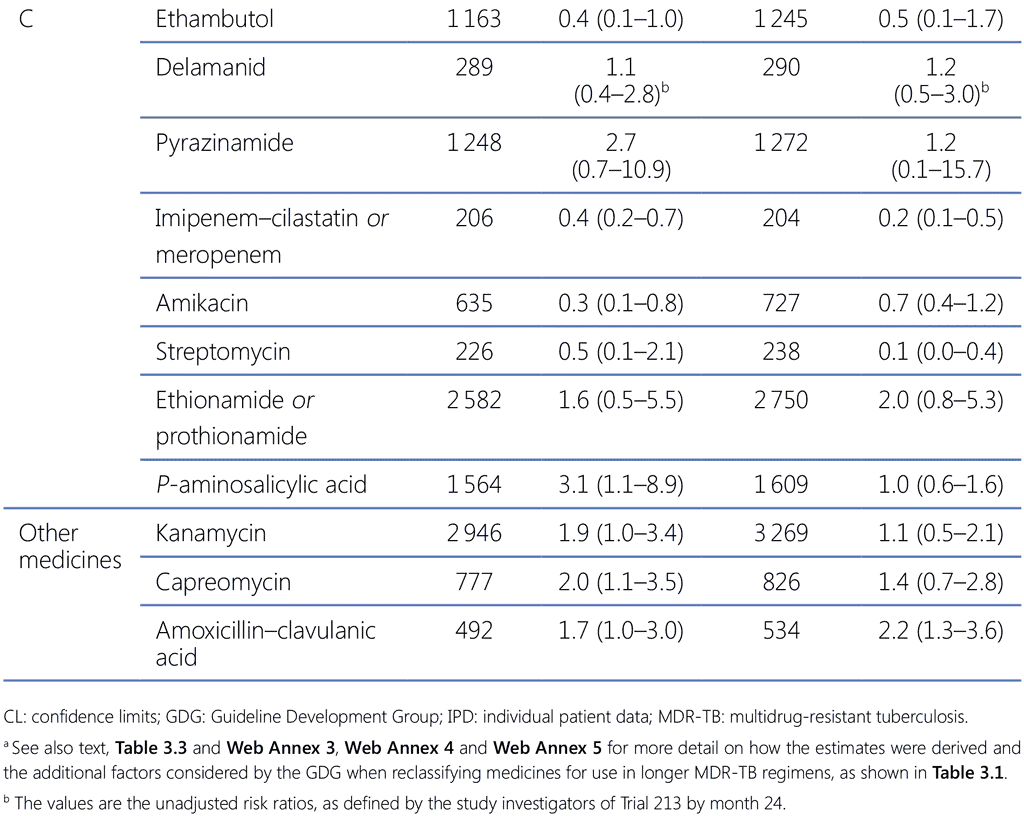
Cuadro 3.2. Riesgo relativo de fracaso del tratamiento o recaída y de muerte (en comparación con el éxito del tratamiento), metanálisis del conjunto de datos de pacientes individuales del 2018 de esquemas alargados de tratamiento contra la TB-MDR y ensayo 213 sobre el delamanid (población del análisis por intención de tratar)a
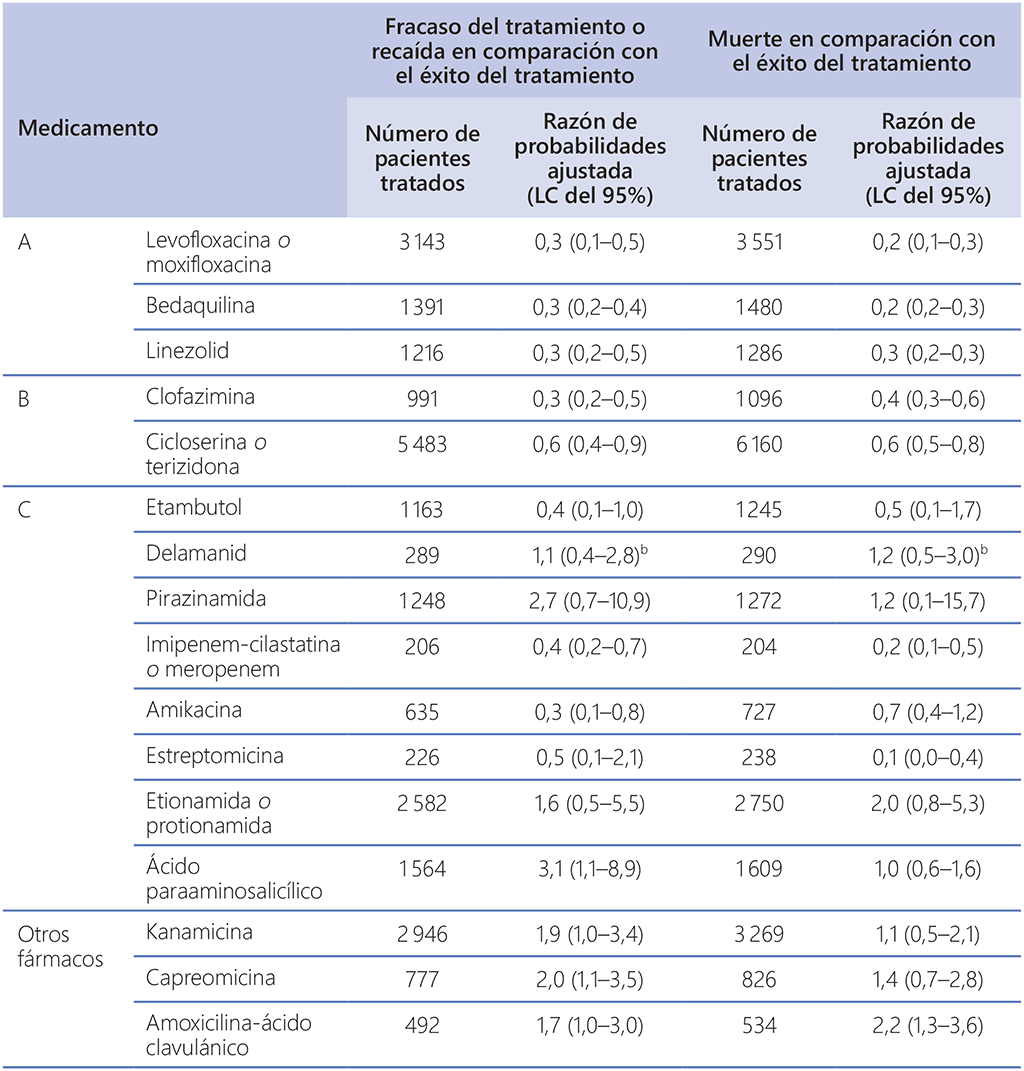
LC: límites de confianza; TB-MDR: tuberculosis multirresistente.
a Véanse también el texto, el cuadro 3.3 y los anexos 3, 4 y 5 en la web para obtener más información sobre el modo en que se realizaron las estimaciones y los factores adicionales que tuvo en cuenta el grupo de elaboración de las directrices al reclasificar los fármacos para su uso en esquemas alargados contra la TB-MDR, según se muestra en el cuadro 3.1.
b Los valores son las razones de probabilidades no ajustadas, tal como las definieron los investigadores del ensayo 213 en el mes 24.
Pregunta PICO 4-2018 (TB-RR/MDR, 2018) (número de fármacos que probablemente sean eficaces)
En lo que respecta a la pregunta PICO 4-2018 (TB-RR/MDR, 2018), el análisis mostró que en los esquemas alargados de tratamiento de la TB-MDR, el riesgo de fracaso del tratamiento, recaída y muerte era comparable cuando el tratamiento comenzaba con cuatro, cinco o seis medicamentos que probablemente eran eficaces. El análisis también mostró que los pacientes que tomaron tres medicamentos en la fase de continuación —la situación que se esperaba al comenzar con cuatro medicamentos y suspender el medicamento inyectable al final de la fase intensiva— no tuvieron peores resultados que los que tomaron cuatro medicamentos en la fase de continuación.
Dado que las interacciones farmacológicas, la cantidad de comprimidos o cápsulas que debe tomar el paciente y la probabilidad de eventos adversos aumentan con el número de medicamentos que integran un esquema, sería deseable prescribir a los pacientes el número mínimo de medicamentos necesarios para obtener niveles comparables de curación sin recaídas. Al decidir sobre el número mínimo de fármacos que se debía recomendar, el grupo de elaboración de las directrices del 2018 consideró también algunos análisis que incluían medicamentos inyectables en los esquemas de tratamiento, si bien era plenamente consciente de que, según se prevé, en el futuro habrá cada vez menos medicamentos inyectables en los esquemas alargados. Además, era importante prever situaciones en las que se suspendiera más de un medicamento en algún momento del tratamiento, ya sea por su indicación de uso —el uso de la bedaquilina y del delamanid para indicaciones autorizadas es de 6 meses— o por cuestiones de tolerabilidad (en particular en el caso del linezolid; cuadro 3.3) (66); así pues, durante la mayor parte del ciclo de tratamiento, el esquema contendría dos fármacos clave menos que al inicio. Si bien el uso de la bedaquilina durante más de 6 meses se considera un uso extra en indicación no autorizada, el grupo de elaboración de las directrices del 2019 contó con nueva evidencia sobre el perfil de seguridad del uso de bedaquilina durante más de 6 meses. Esta evidencia confirma que es seguro usar la bedaquilina durante más de 6 meses en pacientes sometidos a programas apropiados de vigilancia al inicio y durante el seguimiento. El uso de la bedaquilina durante más de 6 meses sigue siendo una indicación no autorizada, por lo que siguen siendo pertinentes las prácticas óptimas al utilizarla en tales indicaciones.
El conjunto de datos de pacientes individuales del 2018 incluyó los resultados observados en más de 300 pacientes tratados con linezolid durante al menos 1 mes, la mayoría con una dosis de 600 mg al día, con información sobre la duración de la administración. Alrededor del 30% solo recibieron el linezolid durante 1-6 meses, pero más del 30% lo recibieron durante más de 18 meses, y estos pacientes tuvieron la frecuencia más baja de fracaso del tratamiento, pérdida de contacto durante el seguimiento y muerte. Un gráfico de la duración del tratamiento con linezolid y del fracaso del tratamiento indica que la duración óptima de uso sería de unos 20 meses, lo que corresponde a la duración total habitual de un esquema alargado contra la TB-MDR. Sin embargo, un análisis así no tiene en cuenta el sesgo de supervivencia, según el cual es más probable que los pacientes que completan todo el tratamiento tengan un resultado satisfactorio, dado que las muertes y las pérdidas de contacto durante el seguimiento se producen antes. No se pudo observar un patrón claro del tipo de eventos adversos y de la duración del uso, aunque se notificaron unos cuantos casos de neuropatía óptica asociada al uso del linezolid a largo plazo (67) y se notificó la aparición de toxicidad hemática independientemente de la duración del uso.
Cuadro 3.3. Eventos adversos graves en pacientes que siguen esquemas alargados de tratamiento contra la TB MDRᵃ
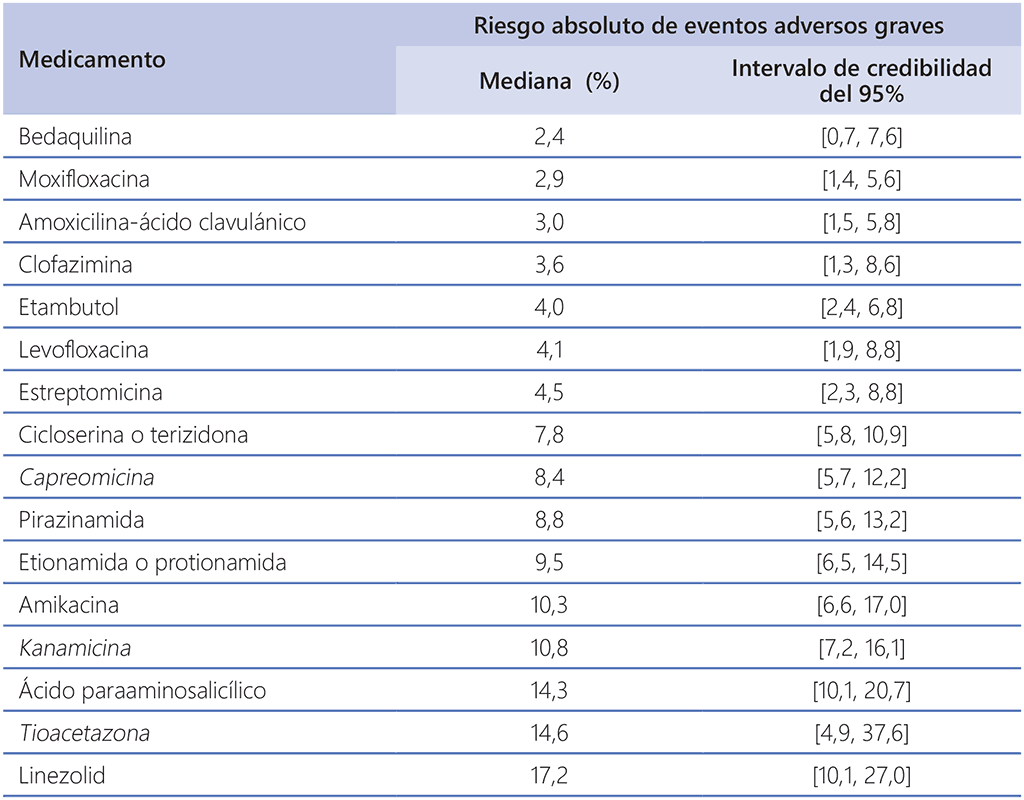
TB-MDR: tuberculosis multirresistente; TB: tuberculosis.
a A partir de un metanálisis “en red basado en grupos” de un subconjunto de pacientes del conjunto de datos de pacientes individuales del 2016 respecto a los cuales se notificaron eventos adversos que motivaron la suspensión permanente de un fármaco contra la TB (27 estudios) o que se clasificaron como de grado 3-5 (3 estudios). Hay ligeras diferencias entre las estimaciones finales citadas en la publicación resultante (66) y los valores calculados en el momento en que se reunió el grupo de elaboración de las directrices y se muestran en este cuadro, ya que en la publicación se utilizó un conjunto de datos ampliado; sin embargo, esas pequeñas diferencias no influyen en las conclusiones extraídas sobre el uso de estos fármacos. No hubo registros suficientes sobre el delamanid, la combinación imipenem-cilastatina y el meropenem para estimar los riesgos. Los fármacos que no están en los grupos A, B o C se destacan en cursiva.
En el 2018, el grupo de elaboración de las directrices recomendó que, cuando sea posible, los esquemas se compongan de los tres medicamentos del grupo A y al menos un medicamento del grupo B, de modo que el tratamiento comience con al menos cuatro medicamentos que probablemente sean eficaces y que al menos tres medicamentos se mantengan durante el resto del tratamiento si se suspende la bedaquilina al cabo de 6 meses. En el 2019, el grupo de elaboración de las directrices contó con nueva evidencia sobre el perfil de seguridad del uso de la bedaquilina durante más de 6 meses. Esta evidencia confirma que es seguro usar la bedaquilina durante más de 6 meses en pacientes sometidos a programas apropiados de vigilancia al inicio y durante el seguimiento. Si solo se pueden utilizar uno o dos fármacos del grupo A, se deben incluir ambos fármacos del grupo B. Si no se puede configurar un esquema exclusivamente con fármacos de los grupos A y B, se agregan fármacos del grupo C para completarlo. En los pacientes en los que es más probable que se suspendan dos fármacos del grupo A antes de que finalice el tratamiento (por ejemplo, en caso de enfermedades concomitantes preexistentes que exigen suspender antes de lo previsto tanto la bedaquilina como el linezolid debido a riesgos para la salud), puede ser aconsejable comenzar con cinco medicamentos eficaces en lugar de cuatro. Se prevé que estas disposiciones se apliquen a la mayoría de los pacientes con TB-RR/MDR, incluidos los que presentan resistencia adicional a las fluoroquinolonas o a otros fármacos.
Pregunta PICO 8-2019 (TB-RR/MDR, 2019) (uso de la bedaquilina durante más de 6 meses)
En lo que respecta a la pregunta PICO 8-2019 (TB-RR/MDR, 2019), el análisis mostró una ORa de 1,5 (IC del 95%: 0,7, 2,7) para el éxito del tratamiento en comparación con el fracaso del tratamiento; 0,8 (IC del 95%: 0,2, 0,4) para el éxito del tratamiento en comparación con la muerte; 1,0 (IC del 95%: 0,5, 1,7) para el éxito del tratamiento en comparación con el fracaso del tratamiento o la muerte; y 0,8 (IC del 95%: 0,5, 1,2) para el éxito del tratamiento en comparación con el conjunto de todos los resultados desfavorables. Los revisores de la evidencia habían previsto utilizar dos métodos analíticos concebidos para minimizar el sesgo, a saber, modelos estructurales marginales, para tener en cuenta factores de confusión que varían con el tiempo, y el emparejamiento exacto y por índice de propensión de las características de los pacientes. Sin embargo, el tamaño muestral comportó limitaciones en cuanto a la forma de aplicar el primer método; además, debido a las limitaciones del conjunto de datos, los especialistas en bioestadística advirtieron que no era posible ajustar los factores de confusión según el plan original de análisis de datos. El grupo de elaboración de las directrices del 2019 observó que la población incluida en los estudios que se evaluaron estaba muy seleccionada, y que era posible la confusión por indicación (es decir, era probable que la bedaquilina se hubiera administrado durante más de 6 meses a las personas en quienes había factores clínicos por los cuales estaba indicado un tratamiento prolongado con bedaquilina). El grupo de elaboración de las directrices del 2019 concluyó que era sumamente probable la confusión residual en los datos, y que la población de pacientes comprendida en el estudio no permitiera extrapolar los resultados al uso corriente en todos los pacientes con TB-RR/MDR. Ello impidió que se formulara una recomendación formal sobre la eficacia o la efectividad del uso de la bedaquilina durante más de 6 meses; no obstante, el grupo de elaboración de las directrices del 2019 llegó a la conclusión de que se podía hacer una declaración sobre su seguridad. Esta información se presenta en la sección 3.5 y en una nota del cuadro 3.1.
En cuanto a los eventos adversos, de los 750 pacientes que recibieron bedaquilina sin administración simultánea de delamanid en el estudio de observación endTB (exposición total de 6316 meses-persona), 26 pacientes presentaron un evento adverso relacionado con el fármaco (tasa: 0,44 por cada 100 meses-persona de exposición); en 16 de ellos, se clasificó como un evento adverso grave (tasa: 0,25 por cada 100 meses-persona de exposición). Veinte de los 26 eventos adversos relacionados con el fármaco, y 15 de los 16 eventos adversos graves, tuvieron lugar en los primeros 203 días de exposición a la bedaquilina (exposición total de 4304 meses-persona); los restantes (6 de los 26 eventos adversos relacionados con el fármaco y 1 de los 16 eventos adversos graves) fueron posteriores. Ninguno de los pacientes que recibieron bedaquilina durante más de 203 días presentó un evento adverso relacionado con el fármaco (de cualquier grado) en los primeros 203 días de tratamiento. Además, las tasas de eventos adversos relacionados con los fármacos del tratamiento parecieron ser más bajas después de los primeros 203 días: 0,51 en los primeros 203 días en comparación con 0,30 en los días posteriores por cada 100 meses-persona. Asimismo, las tasas de eventos adversos graves relacionados con el fármaco parecieron ser más bajas después de los primeros 203 días: 0,35 en los primeros 203 días en comparación con 0,05 en los días posteriores por cada 100 meses-persona.
Los valores del intervalo QTcF en las personas que recibieron bedaquilina aumentaron en promedio 22 ms (de 397 ms a 419 ms) entre la medición obtenida antes o en el momento de la primera dosis de bedaquilina y la del final del primer mes. En los meses posteriores de exposición, todos los valores de la media del QTcF fueron inferiores a los del final del primer mes (intervalo: 404-419 ms). En cerca del 12% de los pacientes, el QTcF aumentó en más de 60 ms con respecto a los valores iniciales. La prolongación del QTcF en más de 500 ms fue poco frecuente, y se registró en el 0,4-1,5% de los pacientes durante cada uno de los primeros 9 meses, pero no a partir de entonces. El mayor número de casos de QTcF superior a 500 ms se observó en las personas que recibieron bedaquilina y clofazimina; sin embargo, esta fue también la combinación más común de fármacos administrados.
Veintidós personas presentaron eventos adversos cardíacos relacionados con los fármacos; 15 de ellas recibían bedaquilina con clofazimina, pero no moxifloxacina ni delamanid (tasa: 0,3 por cada 100 meses-persona), 5 de ellas recibían bedaquilina con clofazimina y moxifloxacina, pero no delamanid (tasa: 0,3 por cada 100 meses-persona) y 2 recibían bedaquilina y delamanid, independientemente del uso de clofazimina y moxifloxacina (tasa: 0,2 por cada 100 meses-persona). Las personas que recibieron bedaquilina sin clofazimina, moxifloxacina ni delamanid no presentaron eventos adversos.
Con respecto a la exposición a la bedaquilina durante el embarazo, los resultados del estudio de cohortes no mostraron diferencias estadísticamente significativas en los resultados del parto o del embarazo cuando se comparó a los lactantes que habían tenido exposición intrauterina a la bedaquilina con los que no habían estado expuestos (p = 0,741 para los resultados del parto y p = 0,312 para los resultados del embarazo) (51). Hubo 45 nacidos vivos (92% del total) en el grupo expuesto a la bedaquilina y 54 nacidos vivos (90% del total) en el grupo no expuesto. Además, hubo 4 muertes fetales o neonatales en el grupo expuesto a la bedaquilina (8% del total del grupo expuesto a la bedaquilina, con 3 mortinatos y 1 interrupción del embarazo) y 6 muertes fetales y neonatales en el grupo no expuesto a la bedaquilina (10% del total del grupo no expuesto, a saber, 3 mortinatos y 3 abortos espontáneos) (51). Las observaciones del estudio también indicaron que los resultados del tratamiento fueron más favorables en las embarazadas expuestas a la bedaquilina en comparación con las no expuestas (el 71% en comparación con el 62%, respectivamente, p = 0,349) (51). Los resultados del embarazo comprendieron tanto los de nacidos vivos como los de desenlaces desfavorables del embarazo (muertes fetales y neonatales, nacimientos prematuros de menos de 37 semanas y peso bajo al nacer inferior a 2500 g); y los resultados del lactante comprendieron el aumento de peso y los hitos del desarrollo, así como el diagnóstico de TB (51). De todos los resultados del embarazo y del lactante evaluados, solo el peso bajo al nacer se asoció con la exposición intrauterina a la bedaquilina (45% en comparación con el 26%, p = 0,034). El peso medio de los neonatos expuestos a la bedaquilina fue de 2690 g, en comparación con 2900 g en los neonatos que no estuvieron expuestos a la bedaquilina. Sin embargo, no fue posible atribuir de manera concluyente este efecto a la bedaquilina, y serán necesarias nuevas investigaciones para explorar esta relación (51). No hubo diferencias significativas en el crecimiento de los lactantes después del nacimiento: en un subanálisis de 86 lactantes sometidos a seguimiento prospectivo (41 con exposición intrauterina a la bedaquilina y 45 no expuestos), el 88% de los lactantes con exposición intrauterina a la bedaquilina presentaron un aumento de peso normal al año de edad, en comparación con el 82% de los lactantes no expuestos (p = 0,914) (51).
Pregunta PICO 9-2019 (TB-RR/MDR, 2019) (uso simultáneo de bedaquilina y delamanid)
En lo que respecta a la pregunta PICO 9 (TB-RR/MDR, 2019), los análisis mostraron una ORa de 1,6 (IC del 95%: 0,5, 5,4) para el éxito del tratamiento frente al fracaso del tratamiento; 0,8 (IC del 95%: 0,3, 2,1) para el éxito del tratamiento frente a la muerte; 1,2 (IC del 95%: 0,6, 2,5) para el éxito del tratamiento frente al fracaso del tratamiento o la muerte; y 0,6 (IC del 95%: 0,3, 1,1) para el éxito del tratamiento frente al conjunto de todos los resultados desfavorables. En cuanto a los eventos adversos, en los 92 pacientes que recibieron bedaquilina con delamanid simultáneamente durante el tratamiento en el estudio de observación endTB (exposición total de 1095 meses-persona), se produjeron dos eventos adversos relacionados con la bedaquilina y el delamanid (tasa combinada: 0,46 por 100 meses-persona de exposición). Esta tasa fue comparable a las observadas en las personas que recibieron la bedaquilina sola (0,41 por 100 meses-persona de exposición) o el delamanid solo (0,68 por 100 mese-persona de exposición). En los 92 pacientes que recibieron bedaquilina y delamanid simultáneamente se registraron dos eventos adversos graves relacionados con los fármacos, uno atribuido a cada uno de ellos (tasa combinada: 0,09 por 100 meses-persona de exposición). La tasa de esos eventos fue inferior a las tasas de eventos adversos graves relacionados con los fármacos observadas en los pacientes que recibieron alguno de los dos fármacos solo (bedaquilina: 0,28; delamanid: 0,39). No se registraron eventos mortales relacionados con los fármacos en los pacientes que recibieron bedaquilina y delamanid simultáneamente.
Los valores del intervalo QTcF en las personas que recibieron bedaquilina y delamanid aumentaron en un promedio de 15 ms (de 398 ms a 413 ms) entre la medición obtenida antes o en el momento de la primera administración simultánea de bedaquilina y delamanid y el final del primer mes. En los siguientes meses de exposición, la media de los valores del QTcF fue similar a la del final del primer mes (intervalo: 404-420 ms). La prolongación del intervalo QTcF de más de 500 ms fue inusual y se observó en un solo paciente en el séptimo mes de exposición simultánea. Los eventos adversos cardíacos relacionados con los fármacos fueron poco frecuentes y solo se registraron en 2 de las 92 personas expuestas simultáneamente a la bedaquilina y al delamanid (tasa: 0,2 por cada 100 meses-persona). Solo se registró un evento adverso cardíaco grave relacionado con los fármacos (tasa: 0,1 por 100 meses-persona). En las 92 personas expuestas simultáneamente a la bedaquilina y al delamanid no se registraron eventos cardíacos mortales relacionados con los fármacos.
En total, en el estudio de observación endTB (n = 1094) hubo dos eventos cardíacos mortales relacionados con los fármacos (muertes súbitas atribuibles a la prolongación del intervalo QT), y otro paciente presentó una arritmia cardíaca. Las dos muertes se registraron en pacientes que recibían bedaquilina, clofazimina, capreomicina y ácido paraaminosalicílico (pero no moxifloxacina ni delamanid); ambos pacientes presentaban hipopotasemia. No se incluyó a esos pacientes en el análisis relativo a esta pregunta PICO, dado que no cumplieron los criterios de inclusión según el plan de análisis estadístico predefinido. Sin embargo, al reconocer que las estimaciones de los eventos adversos graves eran absolutas y no relativas, el grupo de expertos consideró que la evidencia adicional era importante para hacer un seguimiento cuidadoso, cuando se disponga de los datos finales del estudio de observación endTB.
El grupo de elaboración de las directrices convino en que no había evidencia suficiente para evaluar la eficacia ni la efectividad del uso simultáneo de bedaquilina y delamanid, dado que solo hubo 84 pacientes en el grupo de intervención y que los datos no permitían realizar un análisis significativo para el factor de comparación secundario (el uso prolongado del delamanid solo), ya que las poblaciones eran demasiado diferentes para permitir el emparejamiento que se suele realizar. Esto impidió que se hiciera una recomendación formal sobre la eficacia o la efectividad del uso simultáneo de la bedaquilina y del delamanid; no obstante, el grupo de elaboración de las directrices llegó a la conclusión de que se podía hacer una declaración sobre su seguridad. Esta información se presenta en la sección 3.5 y en una nota de cuadro 3.1.
En los otros datos presentados del ensayo DELIBERATE se destacó que, en los pacientes asignados aleatoriamente a la bedaquilina (n = 28), al delamanid (n = 27) o ambos fármacos (n = 27), la variación en el intervalo QTcF durante el tratamiento con respecto al valor inicial fue de 11,9 ms, 8,6 ms y 20,7 ms, respectivamente.34 De los 27 pacientes que recibieron ambos medicamentos, 10 (37,0%) presentaron un evento adverso de prolongación del intervalo QT de grado 135 y dos (7,4%) presentaron un evento adverso de prolongación del intervalo QT de grado 2. En el grupo de la bedaquilina, el 32,0% y el 3,6% de los pacientes presentaron eventos adversos de prolongación del intervalo QT de grado 1 y 2; en el grupo del delamanid, estas cifras fueron del 41,0% para los eventos adversos de prolongación del intervalo QT de grado 1 y del 7,4% para los eventos adversos de prolongación del intervalo QT de grado 2. Ningún paciente presentó eventos adversos de prolongación del intervalo QT de grado 3 o 4. Los investigadores del estudio concluyeron que los efectos de prolongación del intervalo QTcF por el uso simultáneo de delamanid y bedaquilina no eran mayores que sus efectos aditivos. El grupo de elaboración de las directrices señaló que los eventos adversos de prolongación del intervalo QT registrados en el ensayo DELIBERATE eran marcadores indirectos de la muerte súbita de origen cardíaco. También observó que la levofloxacina fue la fluoroquinolona preferida en los esquemas administrados a los pacientes en el ensayo DELIBERATE y que se vigiló estrechamente el potasio sérico.
Pregunta PICO 1-2021 (TB en la población infantil, 2021) (uso de la bedaquilina en pacientes con TB-RR/MDR menores de 6 años) y pregunta PICO 2-2021 (TB en la población infantil, 2021) (uso del delamanid en pacientes con TB-RR/MDR menores de 3 años)
En lo que respecta a la pregunta PICO 1-2021 (TB en la población infantil, 2021) y la pregunta PICO 2-2021 (TB en la población infantil, 2021), los detalles de la revisión de la evidencia y las deliberaciones del grupo de elaboración de las directrices pueden consultarse en el módulo 5. Manejo de la tuberculosis en la población infantil y adolescente.
34 Comunicación personal, K Dooley, Johns Hopkins Medicine, noviembre del 2019: para esta afirmación y el resto de este párrafo
35 En el ensayo DELIBERATE, un evento adverso de prolongación del intervalo QT de grado 1 se clasificó como un QTcF absoluto en las siguientes situaciones: >480 ms y ≤500 ms y una variación del QTcF respecto al inicio de >0 ms a ≤30 ms O BIEN un QTcF absoluto ≤480 ms y una variación del QTcF respecto al inicio de >30 ms a ≤60 ms. Un evento adverso de prolongación del intervalo QT de grado 2 se clasificó como un QTcF absoluto en las siguientes situaciones: >480 ms y ≤500 ms y una variación del QTcF respecto al inicio de >30 ms a ≤60 ms O BIEN un QTcF absoluto ≤480 ms y una variación del QTcF respecto al inicio de >60 ms. Un evento adverso de prolongación del intervalo QT de grado 3 se clasificó como un QTcF absoluto en la siguiente situación: >500 ms O BIEN un QTcF absoluto >480 ms y una variación del QTcF respecto al inicio >60 ms. Un evento adverso de QT de grado 4 fue una consecuencia potencialmente mortal, por ejemplo, torsade de pointes (taquicardia helicoidal) u otra disritmia ventricular grave asociada (comunicación personal, K. Dooley, Johns Hopkins Medicine, noviembre del 2019).